NOTE: Refer to Table 1 for recipes of media and solution.
1. Setting up and maintaining an embryo collection cage for live analysis21
- Populate a fresh embryo collection cage with fresh flies.
NOTE: While commercially available collection cages are recommended, collection cages can be easily constructed from empty fly bottles21.- Discard or transfer adult flies from bottles when many pupae begin darkening at stage p14; at 20 °C this occurs approximately 14 days after the eggs have been laid22.
- Allow pupae to hatch and populate the bottles for 12-24 h.
- Transfer fresh flies into a small collection cage using a small funnel over the collection cage opening to prevent flies from escaping. Combine up to 3 bottles to produce a densely populated cage containing 200 to 500 flies.
- Transfer flies into the collection cage by tapping a fly bottle against a hard surface repeatedly (to stun the flies). Then open and invert the fly bottle over the collection cage funnel. Tap the open bottle onto the collection cage a few times to drop flies into the collection cage.
- Close the open fly bottle by quickly setting it down onto its plug. While transferring the open fly bottle from the funnel to its plug, ensure that the bottle opening points downward as flies tend to crawl up away from the source of the tapping.
- Press the funnel against the fly cage opening and tap the cage repeatedly on a hard surface to stun the flies inside. Immediately remove the funnel and secure a fresh grape plate to the fly cage opening. Use labeling tape to secure the grape plate. Repeat this process to combine up to 3 bottles to produce a densely populated collection cage.
NOTE: Alternatively, flies can be anesthetize using CO2 or other fly sleeping agents; however, this will increase the acclimation period in the step 1.2.
- Acclimate the new collection cage for 24-48 h. If CO2 or any other sleeping agents were used, acclimate the collection cage for at least 48 h.
- Replace the grape plate in the collection cage with a fresh one containing yeast paste twice a day, 8-10 h apart.
- Setup a fresh collection cage every 5 days.
2. Collecting and preparing embryos for imaging
- Obtaining embryos with synchronized development
- Exchange the grape plate on the collection cage with a fresh one containing a dab of yeast paste at the center. Allow flies to lay embryos for 1 h. Toss the old grape plate.
NOTE: Flies can fertilize embryos prior to laying them. The first collection plate will contain many of these predeveloped embryos. Allowing flies to dump old embryos first will improve developmental synchronization between embryos. - Exchange the grape plate for a fresh one containing a dab of yeast paste at the center and let flies lay embryos for 10 min. Toss the old grape plate.
NOTE: To avoid slowing down embryonic development on cold plates, warm the grape plate and yeast paste to room temperature prior to use. - Exchange the grape plate with a fresh one (without yeast paste) and save the old grape plate, it has the embryos for imaging. If necessary, continue collecting embryos from this cage by repeating step 2.1.2 and 2.1.3.
- Exchange the grape plate on the collection cage with a fresh one containing a dab of yeast paste at the center. Allow flies to lay embryos for 1 h. Toss the old grape plate.
- Age collect embryos for 30-45 min to image syncytial divisions23.
NOTE: To image the syncytial blastoderm stage in embryonic development, the embryos must be mounted and on the microscope within 90 min of collection. When attempting this protocol for the first time, age embryos 10-20 min in step 2.2 to provide extra time to set up between steps. - Dechorionate embryos with 50% bleach in DI water.
- Transfer aged embryos from step 2.2 into a 140 nm sieve by filling the grape plate with DI water, gently dislodging the embryos with a paint brush, and pouring the water with embryos into the 140 nm sieve (Figure 1A).
NOTE: This can be done over a sink or 1,000 mL beaker. - Vigorously rinse embryos with a DI water using a squirt bottle. Dry embryos by blotting the sieve on a dry paper towel. Blot the sieve until it stops leaving water marks on the paper towel (Figure 1B).
NOTE: Remove all yeast paste from embryos as it can render bleach treatment ineffective in the next step. - Fill an empty 10 cm plate with freshly prepared 50% bleach diluted with DI water (1:1 bleach: DI water). Dip the sieve into 50% bleach for 2 min 45 s. Start a stopwatch as soon as the sieve touches the 50% bleach. Blot the sieve in and out of the 50% bleach 3-4x in the first 15 s to breakup embryo clumps (Figure 1C).
- Vigorously rinse embryos with DI water using a squirt bottle. Dry the embryos by blotting the sieve on a dry paper towel. Repeat vigorous rinse and blot drying process for 5x (Figure 1D).
NOTE: This is the most important step for removing left over chorion. Use a full DI squirt bottle and use half of the bottle in this step. - Transfer the blot dried embryos from the sieve to a 10 cm grape plates using a paint brush21 (Figure 1E)
NOTE: Manual dichlorination can be used as an alternative to bleach decorination24.
- Transfer aged embryos from step 2.2 into a 140 nm sieve by filling the grape plate with DI water, gently dislodging the embryos with a paint brush, and pouring the water with embryos into the 140 nm sieve (Figure 1A).
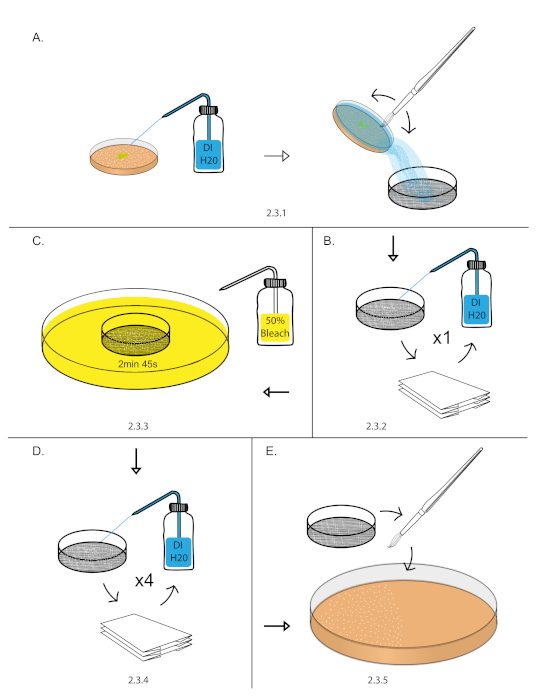
Figure 1: Embryo dechorionation. (A) Embryos were collected using a DI H20 squirt bottle, paint brush, and 140 nm sieve. (B) Embryos were rinsed vigorously using a DI H20 squirt bottle and blot dry over a paper towel. (C) Dechorionation of embryos was performed in 50% bleach for 2 min and 45 s. (D) Embryos were rinsed vigorously using a DI H20 squirt bottle and blot dry over a paper towel. This was repeated 4 times to remove excess chorion. (E) Embryos were then transferred to 10 cm grape plate using a paint brush. Please click here to view a larger version of this figure.
3. Mounting embryos to coverslips
- Under a stereomicroscope equipped with overhead goose neck lighting, organize two rows of 15-20 embryos on a clean section of grape plate using an egg picker tool. Align the embryos on their ventral side in the same orientation with respect to anterior and posteriors ends. To make egg pickers bend fine tip stainless steel tweezers to 45° angle. To review D/V embryos orientation see Campos-Ortega et al.20.
NOTE: It is important for the ventral surface to sit on the grape plate as the embryos will be pressed onto a cover slip in step 3.5, exposing the dorsal surface for imaging. Since the dorsal surface is much flatter than the ventral surface, imaging the dorsal side surface increases the number of nuclei present within a single Z plane. - Prepare 75 mm x 25 mm silicone spacer coverslip.
NOTE: This can be prepared ahead of time. Prepare several coverslips and store them in a slide box to keep them clean.- Trace the outline of a 75 mm x 25 mm coverslip onto a 1.6 mm thick silicone spacer sheet and cut out the silicone spacer using scissors (Figure 2A).
- Using a razor blade, cut out a 40 mm x 15 mm inset from the spacer (Figure 2A).
- Adhere the spacer onto the coverslip and streak 10 µL of heptane glue down the exposed coverslip glass inset (Figure 2A).
NOTE: Putting down and even streak will ensure imaging embryos on the same z plane.
- While the heptane glue from step 3.2.3 dries, cutout a rectangular section of grape plate containing the embryos from step 3.1.
- Use a razor blade to cut a rectangle (smaller than 40 mm x 15 mm) around the two rows of embryos. Do not remove the rectangle yet.
- Cut out a second rectangle next to the first. Remove the second rectangle using the straight edge side of a stainless-steel spatula.
- Carefully slide the straight edge of a stainless-steel spatula under the first rectangle and remove it (Figure 2B).
- Place the grape plate cutout with embryos on the external side of a 3.5 cm dish (Figure 2C).
- Under a stereomicroscope, lower a prepared coverslip over the embryos and gently press the two rows of embryos onto the glue (Figure 2D).
- If microinjecting embryos, skip to step 4.1. If not microinjecting continue to step 3.7.
- Cover embryos by filling the silicone spacer (halfway to the top) with 1:1 – halocarbon oil 700: halocarbon oil 27.
- Line the bottom edges of a 4-slide plate adapter with double sided tape to prevent the cover slip from moving but ensure not to place the tape in the path of the objective lens. This tape is critical. If the coverslip is only sitting on the adapter, it will move throughout the acquisition. Mount the cover slip on the 4 slide plate adapter.
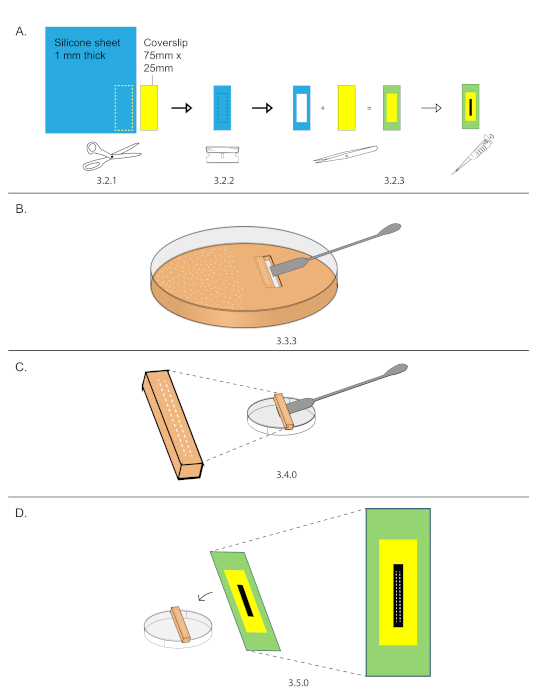
Figure 2: Coverslip preparation and mounting. (A) An outline of 75 mm x 25 mm coverslip was traced onto a 1.6 mm thick silicone sheet. Use scissors to cut out the outline. 40 mm x 15 mm inset were cut out from the silicone spacer and then mounted onto the coverslip. Streaked 15 µL down the center of the inset. (B) Two rows of 15-20 embryos were organized on a grape plate in an area smaller than 40 mm x 15 mm, then that area was cut out using a razor and stainless-steel spatula. (C) Grape plate cut out were placed on the top of an empty 3.5 cm dish. (D) Under a stereomicroscope, the coverslip from (A) was lowered and the embryos were stuck onto the streak of glue. Please click here to view a larger version of this figure.
4. Desiccating and microinjecting embryos
- Place the coverslip with embryos over a slide to prevent the bottom of the coverslip from becoming dirty during desiccation and microinjection.
NOTE: The cover slip will be imaged without a slide attached to it. Therefore, an inverted microscope is necessary for imaging. - Desiccate embryos for 5-10 min, depending on the temperature of the room. The embryos used for imaging in the representative results were desiccated for 7 min at 20 °C in a chamber containing a 1:1 layer of silica gel beads over a drying agent (Figure 3A).
- Immediately after desiccation, cover embryos with 1:1 – halocarbon oil 700: halocarbon oil 27. Fill the silicone spacer halfway to the top.
NOTE: It is important to mix a high viscosity halocarbon oil with a low viscosity halocarbon oil. Pure halocarbon oil 700 (high viscosity) slows down cell cycles progression 30 min post application. A 1:1 mixture of halocarbon oil 700 and halocarbon oil 27 fixed this artifact. - Load microinjection needles with injectant, then mount one to the microinjector and cut it open under pressure.
NOTE: Practice step 4.4 a few times before performing the full assay with any valuable reagents. This is the most difficult step in the microinjection process.- Load microinjection needles with 2 μL of injectant using an extended loading tip. Only one microinjection needle is required, but it is good to have spare loaded needles close by so prepare 2 or 3 needles.
NOTE: Refer to the Table of Materials for specifications on extended loading tips. Microinjection needles cannot be loaded without extended loading tips due to the unique nature of our microinjection technique, which relies on building air pressure inside of a sealed and loaded needle prior to opening its tip. - Mount a needle onto the microinjector and depress the plunger halfway to the tip. The goal is to increase air pressure inside of the glass capillary (Figure 4A).
NOTE: Use a plunger mineral oil based microinjector without mineral oil. Instead of building pressure with mineral oil, use the plunger to build air pressure. - Lower the glass needle onto a glass slide and cut the tip of the needle open using a new #9 razor. Cut the needle at a 45° angle to produce a sharp open tip. The injectant should flow down to the bottom of the tip without flowing out of the needle. Carefully add 20 μL of halocarbon oil over the tip of the needle and re-cut it open at 45°. The injectant should begin to flow rapidly.
- Immediately lower air pressure by retracting the plunger but ensure to maintain a continuous flow rate to prevent the needle from clogging with embryo membrane between injections. (Figure 4B).
NOTE: If the injectant is clear, view the injectant flow rate by lifting the microinjection needle in and out of halocarbon oil and observing the rate of formation for injectant droplets under a stereomicroscope. Adjust air pressure by depressing the plunger a few clicks at a time, then dip the microinjection tip in and out of halocarbon oil to view the rate of formation for new droplets post adjustment. - Use the micromanipulator to position the needle at the center of the field of view, then lift the needle using the micromanipulator and remove the glass slide from under it. The needle is now calibrated for injection.
- Load microinjection needles with 2 μL of injectant using an extended loading tip. Only one microinjection needle is required, but it is good to have spare loaded needles close by so prepare 2 or 3 needles.

Figure 3: Desiccation chamber diagram. (A) This is a diagram of a desiccation chamber. The chamber consists of a 1:1 layer of silica gel beads over a layer of drierite. To perform desiccation, embryos were placed on top of a single well plate lid. The desiccation chamber was closed 7 min. Please click here to view a larger version of this figure.
- Microinject embryos
- Place the embryos on the stereomicroscope stage under microinjection needle but do not lower the needle.
- Focus on embryos at 50x magnification.
- Lower the needle until it comes into the same focus plane as the embryos. Moving the slide with one hand and operating the microinjector with the other, inject one row of embryos.
- Raise the needle and rotate the slide 180° to expose the second row of embryos to the microinjection needle. Lower the needle and inject the second row of embryos.
NOTE: Try to do this in under 10 min. Leave some un-injected embryos to be used as controls.
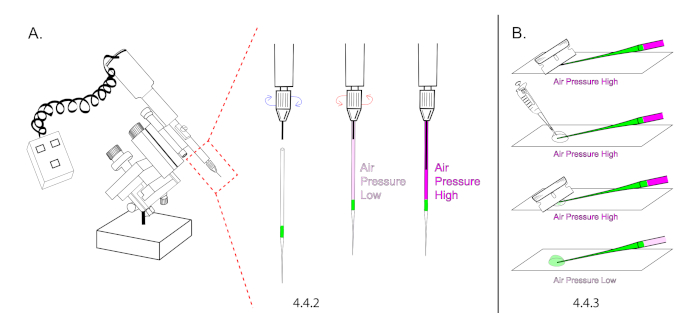
Figure 4: Microinjection needle preparation. (A) In this illustration, the intensity of magenta in needle is representative for air pressure. A loaded needle was mounted onto the microinjector and the plunger was extended 50% to increase air pressure inside of the needle. Make sure the plunger is maximally retracted prior to mounting. (B) The needle tip was cut under pressure and the plunger was retracted to decrease air pressure and the flow rate. Please click here to view a larger version of this figure.
5. Imaging on a high content analyzer
- Prior to running this assay create a plate map for a slide adapter. Use the plate holder editor found in Plate Map Manager menu to enter the dimension for a custom plate adapter.
NOTE: A custom plate map was made for the commercially available plate adapter (see Table of Materials). The plate map contains the following settings; Slide thickness: 1000 µm, Bottom height: 1.607 mm, bottom height variability: 100 µm, substrate material: glass, coverslip thickness: 150 µm, coverslip position: bottom – rows: 1 – spacing: 56.987 mm, columns: 4 – spacing 28.391, size and shape: rectangle, width 23.00 mm, height: 73.00 mm, offset to A1 21.29 mm – 42.74 mm) - Load the slide adapter on the microscope and use the Anteprima feature to tile a brightfield 10x image of the coverslip.
- Select an embryo from the top or bottom of a row, then select Objective and switch to 60x (NA 0.95). Determine an appropriate exposure for the desired fluorescence channel.
NOTE: Maintain the laser intensity under 25% for live cell imaging. - On the main dashboard, use the Anteprima feature to tile a fluorescent 60x image containing both rows of embryos.
- On the main dashboard, select the Binning drop down menu and set the binning to 1 x 1 for optimal resolution.
- Use the z Stack Setup tab to define parameters for z stacks.
- In the z Stack Setup tab, use the Up and Down arrows to find then define top and bottom z stack limits. Once the top and bottom limits are defined, select the Green Pencil Icon to save the z stack position. Note the z stack position as it will be used in step 5.6.5.
- In the z Stack Setup tab, use the z Step input box define spacing between z slices.
NOTE: The total number of slices is determined by modulating step 5.6.1 and 5.6.2. - On the main dashboard, right click on the Fluorescent Channel and use the Image Mode drop down menu to set the imaging mode to 3D.
- On the main dashboard, select the Laser Autofocus Box to disable autofocus.
- On the main dashboard, define the Initial Focus with the z-stack position from step 5.6.1 then select the Initial Focus Box to disable the “Refocus at each Time Point” feature.
- In the Fields tab, use the Field placement drop down menu to enable Custom Points, then use the Arrows to select points for imaging.
NOTE: The time interval between acquisitions (step 5.8) will determine how many custom points can be imaged simultaneously. - In the Time Series tab, select the Acquire Time Series box to enable the time series feature, then define the total number of time points.
NOTE: 6 custom points x 5 z slices x 80 stacks = 2,400 images, 2,400 images x 8.2 MB per image = 19.68 GB disk space. - On the main dashboard, use the Name Input Box to name the acquisition settings then select File and Save to save the acquisition settings.
- On the main dashboard, select the Scan button to run the acquisition.
6. Post image processing
- Drag and drop the xdce file into Fiji to open the image series as a multidimensional hyperstack.
- Save the hyperstack as a tif file.
- Create max intensity projections and save them as a tif files.
- Assemble a library of max intensity projections and build a pipeline for data extraction and analysis.
The approach presented in this protocol was used to examine the role of microtubules in Endoplasmic Reticulum (ER) reorganization during mitosis in the Drosophila embryo15. During mitosis, the structure of the ER displays a dramatic reorganization, however, the forces that drive these changes are poorly understood. Recently, a family of ER shaping proteins were shown to promote the formation of ER tubule25,26,27,28, which are known for their close proximity with microtubules28. To study the roll of microtubules on ER morphology, the methods provided in the protocol were used to generate data to quantitatively compare the effects of several microinjected drug treatments on ER reorganization during mitosis. Colchicine, which prevents new microtubule polymerization, was found to drastically reduce the reorganization of the ER during mitosis as shown in Figure 5 and Figure 6. Furthermore, the approach described here enabled the production of time laps imaging data for 32 embryos. Mean and max intensity measurements were produced from 12,800 regions of interest using a custom MATLAB script, which also generated descriptive and inferential statistics vital for making comparisons between drug treatments. Due to the availability of molecular probes and the ease of microinjections, the methods described here are easily adaptable to study a variety of biological processes via fluorescence intensity quantification.

Figure 5: Representative Images of Rtnl1-GFP and ReepB-GFP enrichment at the spindle poles during mitosis. (A) 60x field of view of ReepB-GFP during mitosis in cell cycle 10. The blue square represents the field of view used for panels B-E. Panel A is processed with a threshold binary filter. This filter was not applied to Panels B-E since it excludes data from pixels below the set threshold. (B,D) ReepB-GFP in control, and in colchicine. (C,E) Rtnl1-GFP in control, and in colchicine. This figure has been modified from Diaz et al.15. Please click here to view a larger version of this figure.
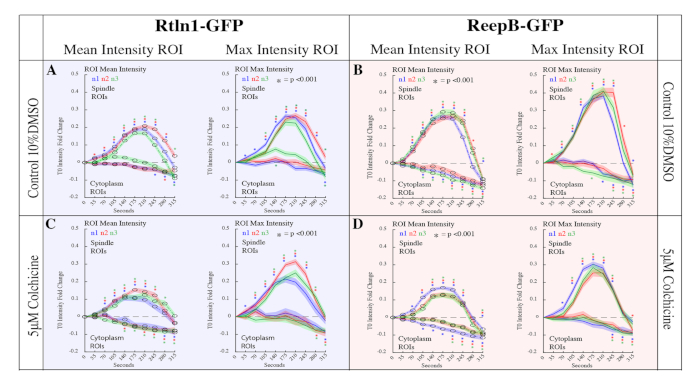
Figure 6: Quantitation of Rtnl1-GFP and ReepB-GFP enrichment at the spindle poles during mitosis. 20 spindles and 20 cytoplasm ROIs across 10 frames were analyzed for 32 embryos in the following conditions. (A) 10% DMSO control injections for Rtnl1-GFP. Control embryos showed a 0.1-fold increase in enrichment (p<0.001 for all samples at T210) compared to non-injected samples (p < 0.001 for all samples at T210). (B) ReepB-GFP embryos injected with 10% DMSO to serve as control. Control embryos showed a 0.1-fold increase in enrichment (p<0.001 for all samples at T210) compared to non-injected samples (Figure 1E; p < 0.001 for all samples at T210). (C) Rtnl1-GFP embryos injected with 5 μM Colchicine. Here we measured a reduction in Rtnl1-GFP enrichment to spindles (p < 0.001 for all samples at T210) compared to controls (A) (p < 0.001 for all samples at T210). (D) ReepB-GFP embryos injected with 5 μM Colchicine. We measured a reduction in ReepB-GFP enrichment to spindles (p<0.001 for all samples at T210) compared to controls (B) (p<0.001 for all samples at T210). This figure has been modified from Diaz et al.15. Please click here to view a larger version of this figure.
| 1. Yeast Paste21 | 2. Grape Plates21 | 3. Heptane Glue24 |
| 1.1 Add 7 grams of active dry yeast to a 50 ml conical. Add 9 milliliters of DI water and mix to even constancy with a metal spatula. | 2.1 Make solution a: Add 6 grams of agar to 140ml of DI water in a 250ml flask and autoclave it. | 3.1 Fill a glass scintillation vial with 50 inches of double-sided tape. |
| 1.1 Add 7 grams of active dry yeast to a 50 ml conical. Add 9 milliliters of DI water and mix to even constancy with a metal spatula. | 2.2 Make solution b: Mix 0.1 grams of methyl paraben in 2ml of ethanol, then add this solution to 60ml of grapefruit juice concentrate. | 3.2 Add 20 ml of heptane and seal glass scintillation vial with parafilm. Rotate overnight at room temperature. |
| 2.3 Immediately after autoclaving solution a, mix in solution b and pour mixture into 10 x 35mm perti dishes. (makes 35 grape plates) | 3.3 Remove plastic tape debris by transferring glue into 2 15 ml conical tubes and centrifuging at 3000 RPM for 15 minutes. | |
| 2.4 Allow grape plates to set at room temperature overnight with plate lids off. Cover plates with lids and store in 4C° after they set. | 3.4 Transfer glue supernatant into 1.5 ml microcentrifuge tubes for storage. |
Table 1: Recipes of media and solutions.
