The details of the reagents and the equipment used in the study are listed in the Table of Materials.
1. Preparation and extrusion of gradient syringes
NOTE: Gradient syringe preparation should be performed using sterile technique.
- Use several colonies of Methylomonas sp. LW13 freshly grown on a plate to inoculate 6 mL of nitrate mineral salts (NMS) medium in an 18 mm x 150 mm glass tube. Seal the tube with a serum stopper and aluminum crimp seal, and add methane using a syringe to a final atmosphere of 50% (v/v) methane in air. Shake this planktonic liquid culture at 200 rpm at room temperature until turbid (about one day).
- Passage liquid cultures 1:10 into fresh media. Continue growing liquid cultures of methanotrophs to log-phase growth (OD600 of ~0.5) and adjust to an OD600 =1.0.
- Prepare syringes by removing the accompanying plunger and keeping them in a sterile container. Attach a sterile PTFE filter tip to the syringe and place it in a standard test tube rack with the tip facing down.
- For each 10 mL syringe, thoroughly mix 1 mL of cells from step 1.2 with 5 mL of NMS and 4 mL of molten agarose (0.5% m/v, cooled to 55 °C) in a sterile conical tube. These volumes can be scaled up to fill multiple syringes in parallel.
- Slowly pour or use a serological pipet to add the mixture to each syringe, up to the 8 mL marking. Allow agarose within syringes to solidify (~15 min), then cap with a sterile 20 mm rubber butyl stopper. Secure the stopper to the syringe using lab tape and label it with syringe contents.
- To add methane to the syringe headspace, fill a large (60 mL) syringe with 100% CH4 and attach a PTFE filter tip (0.2 µm, 25 mm) connected to a sterile needle (23 G). Pierce the rubber stopper with the large syringe and pierce a second sterile needle through the stopper to create a gas outlet.
- Depress the plunger on the large syringe to allow 20 mL of 100% CH4 to flush through the headspace, taking care to remove the outlet needle when there is 1-2 mL of CH4 left in the large syringe to prevent oxygen backflow through the outlet needle.
- Incubate the syringes at 18 °C, repeating steps 1.6 and 1.7 daily to replenish the methane.
- To extrude agarose, replace the PTFE filter tip with a sterile 23 G needle and replace the rubber stopper with the supplied syringe plunger. Slowly depress the plunger to dispense 1 mL increments into separate sterile 1.5 mL microcentrifuge tubes.
2. Determining counter gradient gas concentrations
- Measuring the dissolved oxygen gradient
- Use a razor blade to slice through the width of an agarose-filled syringe (prepared following steps 1.1-1.8) close to the PTFE filter. Fasten the opened syringe to a syringe pump oriented towards a Clark-type microelectrode, with the open end facing the tip of the electrode.
- Adjust the settings of the syringe pump to move the syringe towards the microelectrode at a rate of 1 mL/min (0.6 cm/min); begin recording the dissolved oxygen measurements on the Unisense Logger software as soon as the syringe pump begins moving.
- Measuring the methane gradient
- Immediately before extrusion, replace the syringe filter tip with a one-way stopcock connected to a 23 G needle, and quickly swap the rubber stopper with a syringe plunger. Add eight 1 mL agarose aliquots to separate evacuated 12 mL gas-tight vials and allow the samples to equilibrate at room temperature for 1 h.
- Equilibrate sample vials to atmospheric pressure by cracking open and immediately resealing vials or piercing and quickly removing a needle. Inject 500 µL of vial headspace into a gas chromatograph with flame ionization detection (GC-FID) using a gas-tight syringe. Create a calibration curve derived from CH4 standards to convert peak area (pA*min) to µmol/L.
3. Counting cells in the gradient syringe
- Flow cytometry
- Extrude 1 mL of agarose segments from gradient syringes inoculated with either wild-type or mutant LW13 as outlined in step 1.9. Also, prepare and extrude agarose from a cell-free, sterile syringe as a negative control.
- Add 0.75 mL of 0.85% (m/v) NaCl in water to all extruded agarose samples and homogenize by vortexing. Further, dilute samples 1:10 by transferring 100 µL into a new microcentrifuge tube and adding 900 µL of the salt solution.
- Add 3 µL of a 1:1 mixture of SYTO9 and propidium iodide stains, then incubate in the dark at room temperature for 15 min. To determine the cells per mL of agarose, sonicate the microsphere counting bead suspension in a water bath for 5 min. Then, add 10 µL of the suspension to each sample before flow cytometry analysis.
- Analyze samples with a flow cytometer10,11 with the following parameters: triggering on green fluorescence, flow rate of 10 µL/s, and particle analysis rate below 1,000 particles/s.
- Compare SSC vs. FITC dot plots between cell-free control samples and inoculated agarose samples to draw "bacterial event" voltage gates that exclude background agarose particles. Additionally, draw voltage gates for the microsphere counting beads, which should be consistent between samples.
- To determine the concentration of cells in each agarose segment within the gradient syringe, use the following equation, noting that the dilution factor for the above protocol is 17.7275 and that 10-6 mL is the volume of one microsphere bead.
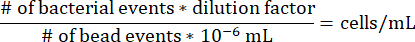
- Counting colony-forming units within the gradient syringe
- Extrude 1 mL of agarose segments into separate, sterile 2 mL microcentrifuge tubes, add 800 µL of NMS, and vortex for 10 s to aid in pipetting.
- Prepare a sterile 96-well plate by adding 180 µL of NMS to each well. Add 20 µL of diluted agarose samples to each well in the first column and pipet to mix.
- Using a multi-channel pipette, serially dilute samples tenfold by transferring 20 µL from the first row of wells into the second row of wells and pipetting 10 times to mix. Continue this process until the last row of the plate.
- Label square grid plates containing NMS agar or media of choice. Using a multi-channel pipette, spot 5 µL from a column of the 96-well plate onto the agar plate. Depending on the size of the agar plate, multiple columns can be spotted on the same plate.
- Incubate plates under 40% methane in air and grow at 18 °C. Count bacterial colonies after 2-3 days and determine the colony-forming units per milliliter (CFU/mL).
4. Biomolecule detection assays
- Polysaccharide assay
- Extrude 1 mL of agarose segments into separate 2 mL microcentrifuge tubes and mix with 1 mL of a 1% (m/v) Na2CO3 solution in water. Heat samples to 80 °C for 30 min with vortexing every 5-10 min, followed by centrifugation at 4,000 x g at 4 °C for 20 min.
- Collect the supernatant combined with three volumes of 100% ethanol and incubate at 20 °C for at least 2 h (or overnight).
- Collect the ethanol-precipitated polysaccharides by centrifugation at 16,100 x g at 4 °C for 30 min. Remove the supernatant and air-dry the pellet. Resuspend the pellet in 100 µL of deionized water.
- Measure the relative polysaccharide content of each agarose segment using a phenol-sulfuric acid colorimetric assay12,13. Combine 50 µL of the resuspended extract with 150 µL of concentrated sulfuric acid and 30 µL of 5% (v/v) phenol in water in a clear 96-well plate.
- Measure the absorbance at 490 nm using a microplate reader and calculate the relative polysaccharide content of each agarose segment as a percentage of the absorbance of the agarose segment closest to the PTFE filter.
- Protein assay
- Extrude agarose into separate 1.5 mL microcentrifuge tubes and transfer 100 µL of each aliquot to glass test tubes.
- Determine the total protein concentration using the test tube protocol of a BCA protein assay kit. Prepare albumin BSA standards with agarose extruded from a sterile syringe as the diluent.
- Extracellular DNA assay
- Extrude agarose into separate 1.5 mL microcentrifuge tubes and transfer 20 µL of each to 0.2 mL microcentrifuge tubes.
- Measure DNA concentrations using the commercially available 1x dsDNA high-sensitivity assay kit following the manufacturer's protocol.
5. RNA extraction
- Prepare the extraction buffer by combining the following in 800 mL of RNase-free water: 2.0 g of CTAB, 2.0 g of polyvinylpyrrolidone (PVP 40), 81.8 g of NaCl, 100 mM of Tris-HCl (pH 8.0), and 20 mM of EDTA. Bring up the volume to 1 L and autoclave; store at 4 °C.
- Aliquot the prepared extraction buffer and add 1% (v/v) final concentration of beta-mercaptoethanol just before use. Warm the buffer to 65 °C using a water bath or heat block.
- Divide each gradient syringe into 1 mL sections by extruding agarose into separate RNase-free 2 mL microcentrifuge tubes following the procedure in step 1.9.
- Centrifuge samples at 21,000 x g, 4 °C, for 15 min and discard the supernatant, keeping samples on ice.
- Add 600 µL of pre-warmed extraction buffer to each pelleted 1 mL of extruded agarose. Add approximately 200 µL of zirconia/silica beads and homogenize samples for 3 min at 30 Hz/s using a bead beater, pausing halfway to place samples on ice for 2 min.
- Centrifuge the samples at 15,000 x g, 4 °C, for 2 min to reduce foaming. Extract samples by adding 600 µL of chloroform: isoamyl alcohol (24:1) to tubes and vortexing for 10 s.
- Centrifuge the samples at 15,000 x g, 4 °C, for 8 min. Carefully transfer the upper aqueous phase to a new RNase-free microcentrifuge tube and add 600 µL of chloroform: isoamyl alcohol (24:1) to the transferred upper phase and vortex for 10 s.
- Centrifuge the samples at 15,000 x g, 4 °C, for 8 min and transfer the new upper aqueous phase to a new RNase-free microcentrifuge tube. Add an equal volume of isopropanol to the transferred upper phase and incubate samples for several hours at -20 °C. Optional: samples can be left at -20 °C overnight.
- Collect RNA-containing precipitates by centrifuging samples at 16,100 x g, 4 °C for 30 min.
- Discard the supernatant and wash the pellet with 300 µL of cold 75% (v/v) ethanol made with RNase-free water and centrifuge at 16,100 x g, 4 °C, for 5 min.
- Wash the pellets again following step 5.10.
- After removing the ethanol supernatant, let the pellets air dry for 15 min. Dissolve pellets in 100 µL of RNase-free water.
- Treat samples with DNase I at 37 °C for 30 min following the manufacturer's protocol.
- Inactivate DNase I by adding 300 µL of acid phenol: chloroform: IAA (125:24:1, pH 4.5). Vortex for 10 s and incubate at room temperature for 5 min.
- Centrifuge at 16,100 x g, 4 °C, for 5 min and keep the upper aqueous phase by transferring it to a new RNase-free microcentrifuge tube.
- Optional: Pool RNA from the same segment across all replicate syringes by combining the upper phases into a conical tube.
- Add 1 volume of isopropanol equal to the volume of the RNA-containing upper phase and add 7.5 M of LiCl to a final concentration of 0.8 M, inverting several times to mix.
- Incubate the samples for several hours at -20 °C. Optional: samples can be left at -20 °C overnight.
- Centrifuge the samples at 16,100 x g, 4 °C, for 15 min and carefully discard the supernatant. Wash the RNA-containing pellet twice by adding cold 70% (v/v) ethanol in RNase-free water, centrifuging at 16,100 x g, 4 °C, for 5 min, and removing the supernatant.
- Allow the pellets to air dry at room temperature for 10 min and resuspend in 50 µL of RNase-free water. Store the RNA samples at -80 °C.
- Optional, depending on the RNA quality: Samples can be re-purified using an RNA purification kit to remove small (<200 nt) RNAs.
- To confirm RNA samples do not contain residual DNA, use 1 µL of purified RNA as the template for PCR amplification using universal bacterial 16S rRNA gene primers 27F/1492R. Include two additional PCR reactions containing 1 µL of either genomic DNA (diluted 1:100) or nuclease-free water to serve as the positive and negative controls, respectively.
- Run the products on a 1% agarose TBE gel using gel electrophoresis14.
NOTE: RNA samples with no DNA contamination should result in the absence of a band in the sample lane. If RNA samples show DNA contamination, reprocess the samples starting from step 5.13. RNA is ready for downstream analysis and can optionally be analyzed to quantify its integrity by measuring the RNA Integrity Number (RIN).
Here, the gradient syringe model ecosystem was used to cultivate a single strain (the methanotroph Methylomonas sp. strain LW13) (Figure 1A)6, but it can also be used to enrich for a methane-oxidizing microbial community by direct soil inoculation (Figure 1B). The presence of a methane-oxygen counter gradient was validated by measuring the concentration of methane and oxygen across cell-free and inoculated syringes (Figure 1C). For LW13-inoculated gradient syringes, a counter gradient formed within one day of flushing the syringe, which steepened over three days of incubation. Over the same time period, a horizontal band formed at the same depth at which both gas substrates reached their lowest concentrations (Figure 1A). The steep gas gradient and depletion of methane and oxygen past the depth of the horizontal band showed that LW13 aerobically metabolized methane and produced a phenotype not observed in homogenous planktonic culture. This phenotype is also produced by other methanotrophic bacteria isolated from the same environmental sample as LW136. Strain-dependent variation in the timing and depth of the horizontal band development among different methanotrophic strains suggested that the horizontal band was affected by the specific behavior of each microbe when cultivated in a spatially resolved context6.
The number of cells in the entire agarose plug was measured using flow cytometry and colony counts (CFU/mL) (Figure 2A). This method was used to compare the cell distribution and survival of wild-type LW13 to a mutant strain of LW13 containing a deletion in the fucose 4-O-acetyltransferase (OAT) gene, which was previously shown to influence horizontal band development6. The ΔOAT mutant of LW13 had lower overall growth in the gradient syringe over 6 days compared to the wild-type, an effect that was not observed in homogenous planktonic cultures of the same strains6. The mutant strain did not form the same distinct horizontal band as the LW13 wild type when cultured in the gradient syringe (Figure 2B). Cell numbers and horizontal band appearance were restored to levels similar to the wild type upon gene complementation in the mutant strain. These results demonstrate that the gradient syringe can be used to link genes to specific phenotypes only present in the methane-oxygen counter gradient.
A variety of genetic, chemical, and molecular techniques were adapted for use with bacteria grown within a semi-solid agarose matrix. The gradient syringe model ecosystem can be readily used for standard biomolecular quantification assays with the inclusion of uninoculated agarose as the negative control. The concentration of three different biomolecules commonly found in extracellular polymeric substances and biofilms was measured: polysaccharides, protein, and extracellular DNA15 (Figure 3). In LW13-inoculated syringe segments, the horizontal band had significantly more polysaccharides than other segments, with no significant increase in protein or extracellular DNA.
RNA-seq was used to measure transcriptional differences in LW13 growing at different depths of the syringe. Robust RNA extraction was achieved using a CTAB-based extraction buffer followed by conventional phenol: chloroform extraction and precipitation steps. The results from the RNA-seq analysis were later used to identify genes implicated in the production of the horizontal polysaccharide band. These results indicate that the semi-solid agarose essential for the creation of a spatially resolved model ecosystem does not prevent further biochemical analyses that are generally reserved for planktonic and plate-based cultures.

Figure 1: The gradient syringe model ecosystem. (A) The gradient syringe inoculated with the methanotroph Methylomonas sp. LW13. A distinct horizontal band (arrowhead) develops within two days of flushing the syringe with 100% methane. (B) Close-up photos of gradient syringes inoculated with soil diluted 10-1 and 10-4 and incubated for two weeks. Gradient syringes containing more dilute soil resulted in spherical colonies throughout the agarose, whereas more concentrated soil inocula resulted in a distinct band. (C) Characterization of the methane-oxygen counter gradient in LW13-inoculated and sterile gradient syringes after three days of incubation. The gray bar indicates the range of depths at which the polysaccharide band was located; data show the mean ± SD of three independent experiments with three technical replicates each. Panels (A) and (C) were modified from Beals et al.6. Please click here to view a larger version of this figure.
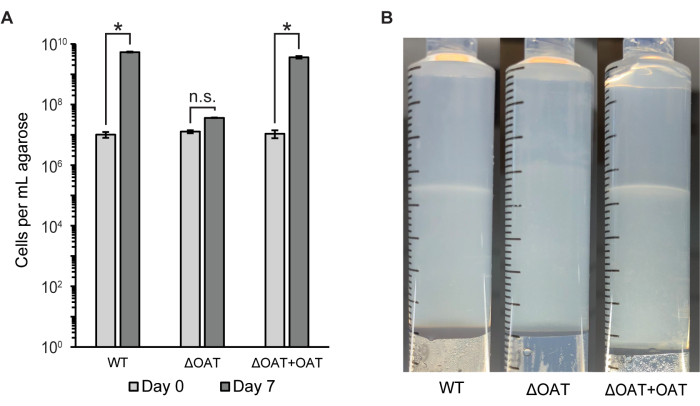
Figure 2: Quantification of LW13 wild type and mutant after incubation in the gradient syringe. (A) The total number of LW13 cells per mL extruded agarose recovered from gradient syringes on Day 0 and Day 7 measured by flow cytometry. ΔOAT contains a deletion of the fucose 4-O-acetyltransferase gene, which was found to be highly expressed in cells located at the depth of the polysaccharide band. ΔOAT+OAT contains the OAT gene inserted at a distal location of the ΔOAT genome. *, significantly different (two-tailed heteroscedastic t-test, α = 0.05); n.s., not significantly different. Data show the mean ± SD of three independent experiments with two technical replicates each. (B) Horizontal band development in gradient syringes inoculated with LW13 wild type, ΔOAT, or ΔOAT+OAT after seven days of incubation. This figure was modified from Beals et al.6. Please click here to view a larger version of this figure.
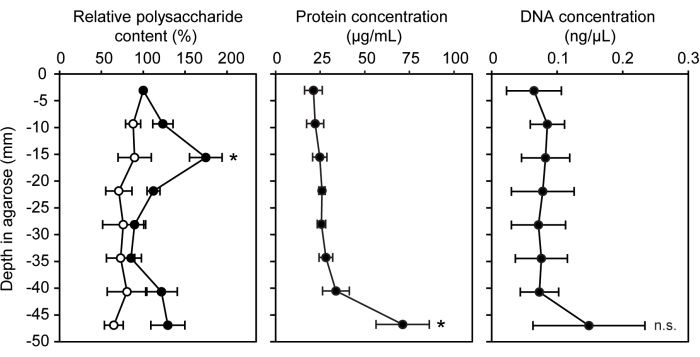
Figure 3: Quantification of biomolecules at increasing depths of the gradient syringe. Relative polysaccharide content (%), protein concentration (µg/mL), and DNA concentration (ng/µL) in eight sections of LW13-inoculated gradient syringes incubated for seven days (filled circles; open circles show values from sterile gradient syringes). Data show the mean ± SD of three independent experiments with three technical replicates each. For relative polysaccharide content, * indicates a significant difference from sterile control at equivalent depth (two-tailed heteroscedastic t-test, α = 0.05). For protein and DNA concentrations, * indicates a significant difference from the section containing the horizontal band (one-way ANOVA with Tukey-Kramer post hoc analysis); n.s., not significant. The figure was modified from Beals et al.6. Please click here to view a larger version of this figure.
