Using the BIBAC-GW system, reporter constructs for studying ODM in plants were generated10. Constructs were designed in the Gateway Entry vector pENTR-gm12 and inserted into pBIBAC-BAR-GW (Figure 1) using the Gateway LR recombination reaction.
Arabidopsis were transformed with pDM19, a BIBAC-BAR-GW plasmid with an mTurquoise-eYFP reporter carrying a translational stop codon in the eYFP reading frame at position 120 (mTurquoise2-eYFP*40) (Figure 2)10. In total, 126 Arabidopsis plants were transformed (9 plants per pot, 14 pots). Seeds of these plants were pooled, sown on trays with soil, and allowed to grow for two weeks prior to treatment with Glufosinate-ammonium solution. Only seedlings expressing the bar gene (present in BIBAC-BAR-GW) survive Glufosinate-ammonium treatment (Figure 3). In total, 11 transgenics transformed with pDM19 were identified, corresponding to a transformation efficiency of 0.02% of the seeds analyzed.
For the 11 transgenics isolated, DNA blotting was used to determine the number of T-DNA integrations. For that purpose, genomic DNA was cut with either BglII or ScaI (strategy as devised on Figure 4A). Both of these restriction enzymes cut only once into the T-DNA sequence (Figure 7A). Hybridization with probes recognizing the bar and eYFP coding regions allowed detection of the number of respective DNA fragments.
The number of individual DNA fragments on the blots allowed for estimating the number of T-DNA insertions in the reporter lines (Table 1). Single hybridizing fragments with both the Bar and eYFP probe indicated the presence of a single T-DNA integration. From the 11 transgenics analyzed, six carried single integrations. The average number of integrations was 1.2.
For 6 lines carrying a single T-DNA integration, the integrity of the inserted reporter construct was tested using DNA blotting (strategy as devised on Figure 4B). Genomic DNA was cut with BglII and PciI to release a 5.5 kb fragment containing both the Bar and mTurquoise-eYFP fusion gene (Figure 7A). A probe against eYFP was used to detect the expected fragment. All plants tested carried an intact fragment. Note that the fragment examined excludes the Left and the Right T-DNA border, and therefore does not examine the integrity of the entire T-DNA, but only the part containing the transgenes of interest.
The expression of the fluorescent reporter gene was determined in independent single-copy transgenic lines differing only by the genomic location of the T-DNA. Relative transcript levels of the CaMV-35S promoter-driven mTurquoise-eYFP reporter were measured by RT-qPCR in four DM19 reporter lines carrying intact, single-copy integrations of which the genomic position was determined10. The variation in reporter gene expression levels between the lines was minor: the maximum difference in mTurquoise-eYFP RNA levels was 2-fold (Figure 8A).
Next, the ODM was carried out in these reporter lines. Three out of the four independent reporter lines showed rather similar ODM efficiencies (Figure 8B). However, one line, DM19[4]1, yielded a very low ODM efficiency compared to the other lines. These results indicate that the ODM is affected by the local genomic context. In what manner the local genomic context of the T-DNA integration in DM19[4]1 differs from that in the other lines remains to be identified. Analysis of available datasets on active and inactive chromatin marks at the genomic T-DNA integration sites in non-transgenic plants did not provide an answer10.
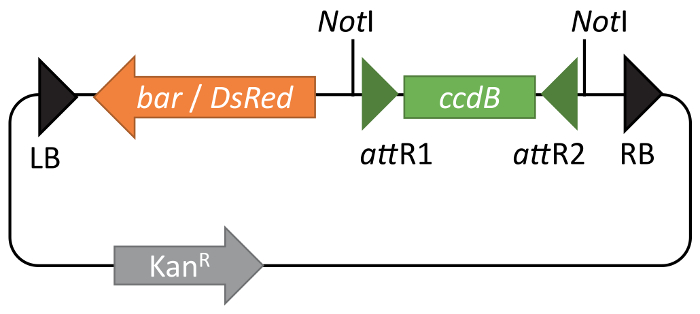
Figure 1: Functional maps of pBIBAC-GW vectors. pBIBAC-GW derivatives are available with either resistance to Glufosinate (bar) or DsRed fluorescence in seed coats (DsRed) as a selection marker in plants. For both vectors, a kanamycin resistance gene is the selection marker in bacteria. The Gateway ccdB cassette is shown between green arrowheads representing recombination sites attR1 and attR2. Please click here to view a larger version of this figure.

Figure 2: Mutagenesis reporter construct. The mTurquoise-eYFP reporter genes are driven by the 35S-CaMV promotor. The mTurquoise coding region is fused to an eYFP coding region carrying a C-A mutation at nucleotide position 120, resulting in a premature translational stop codon TAA, and premature termination of the translation of the fusion protein. The 3′ Nopaline Synthase (3'nos) polyadenylation signal is used to terminate the transcription of the construct22. Nuclear localization signal (NLS) is used to target the translated proteins to the nucleus. Please click here to view a larger version of this figure.

Figure 3: Tray filled with Arabidopsis seedlings before and after Glufosinate-ammonium treatment. Seedlings not expressing the bar gene that is present in the pBIBAC-BAR-GW T-DNA die after being sprayed with Glufosinate-ammonium solution. The photos show the same tray of seedlings (A) before spraying with Glufosinate-ammonium, 14 days after sowing, and (B) 10 days later, after being sprayed twice. Please click here to view a larger version of this figure.
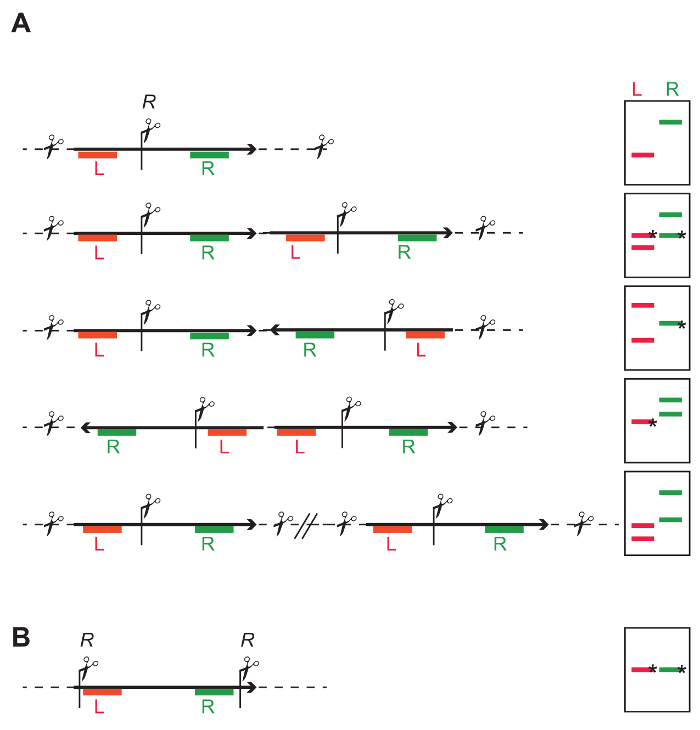
Figure 4: General DNA restriction strategy to identify the number and intactness of inserted T-DNAs. (A) One restriction site (R) in the middle of the T-DNA allows independent probing of the left (red L) and right part of the T-DNA (green R). The cartoons on the right show that depending on single- or multi-copy T-DNA integrations, different banding patterns are obtained with DNA blotting. Bands marked with an * have a defined length, while the length of other bands depends on the closest restriction site in the flanking genomic DNA. Single insert: The L and R probe both give one independent fragment. The expected average fragment size can be calculated based on the frequency of the restriction site in the genome. The minimal size is the distance from the restriction site to the Left Border (LB) or Right Border (RB), depending on which end of the integration is being probed, and if the T-DNA is intact. Tandem repeat: The probes for L and R give both two fragments; for each probe one of the fragments includes flanking genomic DNA, the second fragment has an expected size and is identified by both probes. Inverted repeat: Depending on the directionality of the integrated cassette, either one L and two R fragments, or two L and one R can be identified. Individual single insertions: The result is a number of independent fragments, and the number of fragments corresponds to the number of integrations. (B) Restriction sites at the extremities of the T-DNA allow determining the integrity of the fragment between the restriction sites. Please click here to view a larger version of this figure.
Figure 5: An agarose gel with restriction pattern and the matching transparency. (A) On the agarose gel, genomic DNA digested with EcoRI is shown. The proper digestion of the DNA is illustrated by the presence of discrete satellite bands. (B) Marking the position of the slots and marker bands on a transparency makes it possible to later easily calculate the size of hybridizing fragments. Here, MRC Holland markers (Blue and Red) are used, indicated by M. Please click here to view a larger version of this figure.
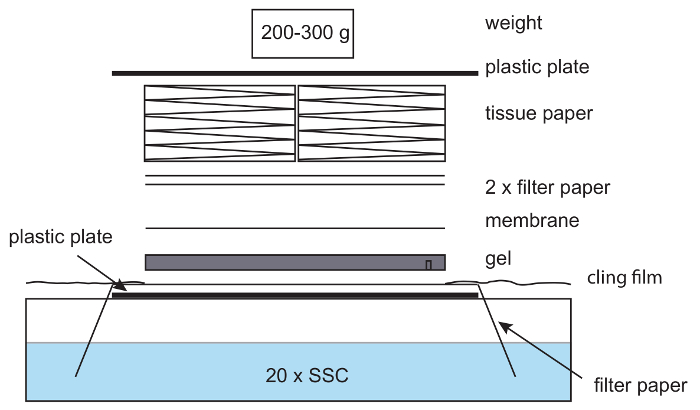
Figure 6: Setup for capillary blotting. In a capillary blotting setup, filter paper is placed on a plastic plate with the ends of the paper hanging in 20x SSC buffer. The paper is wetted with 20x SSC, and an agarose gel placed on top, followed by a nylon membrane, filter paper, and a stack of tissues. A light weight is placed on top. Care is taken to remove air bubbles between the gel, paper and membrane. Cling film is used to avoid drying out of the setup. Please click here to view a larger version of this figure.
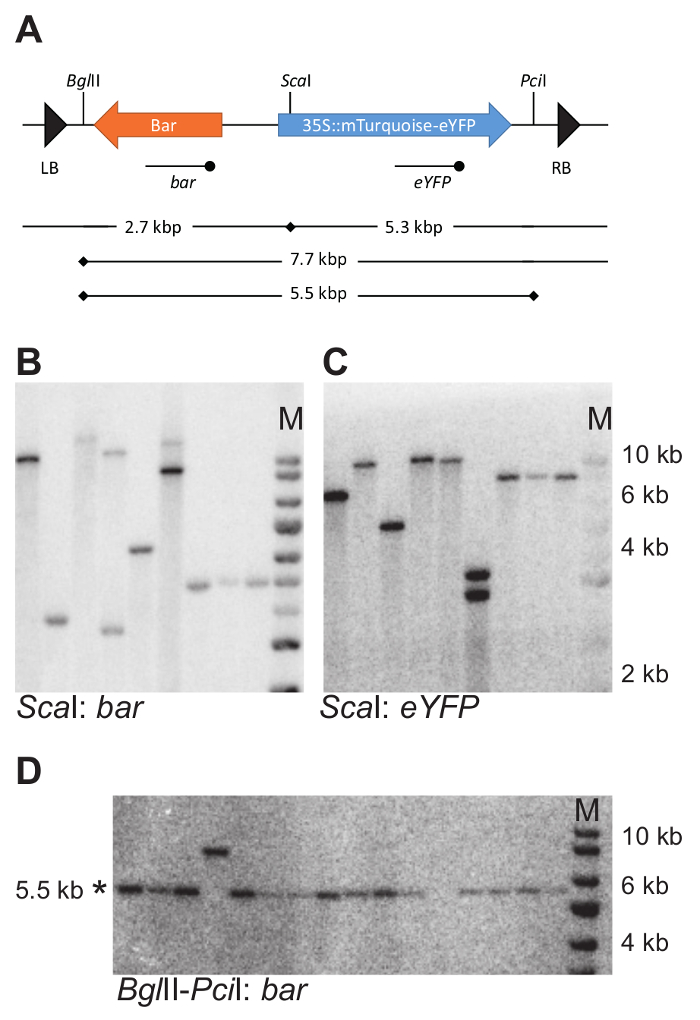
Figure 7: Example of DNA blotting strategy and experimental outcome. (A) DNA blotting strategy to determine the number and intactness of T-DNA integrations. Cutting locations of the selected restriction enzymes within the T-DNA are indicated with vertical bars. The eYFP and bar probes used for hybridization with digested genomic DNA are indicated using a line with the terminal dot below the T-DNA. (B–D) Example DNA blots. Genomic DNA was cut with ScaI and the blot was probed with both a bar and eYFP probe (B and C). Genomic DNA was cut with BglII and PciI and probed with a bar probe. Intact fragments are 5.5 kbp in size (D). Note that the set of samples in D differs from those shown in B and C. * indicates the expected fragment size; M, marker. In B, C, and D the same size marker is used. Please click here to view a larger version of this figure.
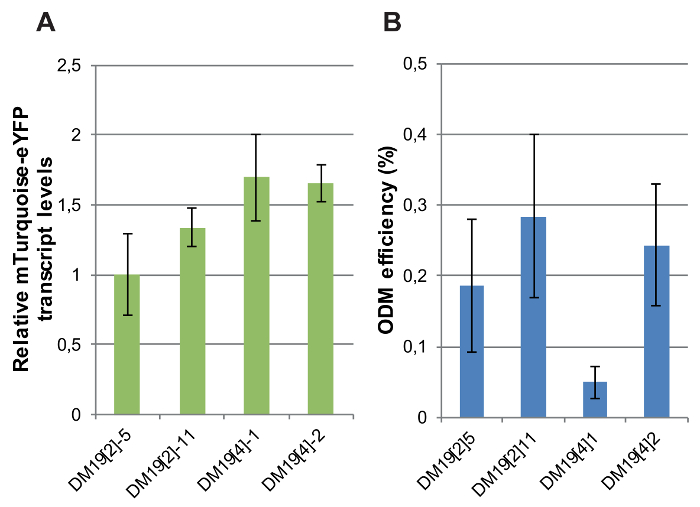
Figure 8: mTurquoise-eYFP expression levels and ODM efficiencies in independent mTurquoise-eYFP reporter lines. (A) Relative mTurquoise-eYFP transcript levels measured by RT-qPCR in DM19 reporter lines. For normalization transcript levels of Actin were used. (B) ODM efficiency measured in the DM19 reporter lines. For A and B, bars indicate the average of at least five biological replicates. Error bars indicate SEM. Please click here to view a larger version of this figure.
| Type of T-DNA locus | Nr of T-DNA integrations | Reporter line | Number of fragments detected | Integrity | |||
| ScaI | BglII | BglII/PciI | |||||
| bar | eYFP | bar | eYFP | eYFP | |||
| Single locus integrations | 1 | 19[2]-2 | 1 | 1 | 1 | 1 | + |
| 1 | 19[2]-5 | 1 | 1 | 1 | 1 | + | |
| 1 | 19[2]-9 | 1 | 1 | 1 | 1 | + | |
| 1 | 19[2]-11 | 1 | 1 | 1 | 1 | + | |
| 1 | 19[4]-1 | 1 | 1 | 1 | 1 | + | |
| 1 | 19[4]-2 | 1 | 1 | 1 | 1 | + | |
| 2, inverted repeat | 19[2]-10 | 2 | 1 | 1 | 1 | + | |
| 2, incomplete integration | 19[2]-3 | 1 | 2 | 1 | 1 | ND | |
| Multiple locus integrations | 2 | 19[2]-6 | 2 | 2 | 2 | ND | |
| 2 | 19[2]-7 | 2 | 2 | 2 | 2 | ND | |
| 3/4 | 19[2]-1 | 4 | 3 | 3 | 3 | ND | |
| ND – not determined. | |||||||
Table 1: Summary of the DNA blotting data for transgenics isolated after transformation with pDM19.
