Confocal analysis of animal cap explants expressing fluorescently tagged proteins provides an effective system for visualising ligand distribution under different experimental conditions. In one example, the distribution of GFP tagged Shh is shown (Figure 1). At the 2-cell stage, Xenopus embryos are injected into both cells with either with a control mRNA or with mRNA coding for Sulf1, an enzyme that modifies cell surface heparan sulfate and influences the Shh morphogen gradient16. These embryos are cultured until the 32-cell stage and a single cell is co-injected with mRNAs coding for Shh-GFP and memRFP (as a lineage tracer). This creates a clone of cells expressing Shh-GFP that is marked with the cell membrane tethered RFP. At the blastula stage, animal cap explants are taken for analysis by confocal microscopy. Figure 1 shows that under control conditions, Shh-GFP is secreted and diffuses outside of the region expressing the injected mRNAs. However, in the presence of Sulf1, Shh-GFP is more restricted in its distribution and, in this sample, is not detected outside the clone of cells producing it. The effects of Sulf1 on Shh-GFP distribution has been analysed more fully16.
When expressed in Xenopus embryos, fluorescently tagged Wnt ligands are secreted, accumulate on the cell membrane, and diffuse across the field of cells. In Figure 2, we characterise the quantitaive and qualitative properties of the two different fluorecently tagged Wnt ligands in cells injected and expressing the proteins. Figure 2A-H shows examples of how Wnt8a and Wnt11b-HA-eGFP accumulate on the membrane of animal cap cells. By comparing panels 2B and 2F it is clear that Wnt8a-HA-eGFP accumulates more efficiently on the cell membrane than Wnt11b-HA-eGFP. Quantitative information has been extracted from the confocal images using a combination of image and script analysis software (supplemental code file). Figure 2I shows the relative accumulation of Wnt8a-HA-eGFP on the cell membrane compared to Wnt11b-HA-eGFP. The data was obtained using a computer script that determines the total number of Wnt-HA-eGFP pixels co-localising with cell membrane pixels in each image. In another approach, the total number of Wnt-HA-eGFP puncta that co-localise with the cell membrane are counted using image analysis software (Figure 2J). Qualitative information about the size and shape of individual puncta can also be extracted from the data using image analysis software. In addition to fewer Wnt11b-HA-eGFP puncta co-localising with the cell membrane (Figure 2J) these puncta also have a smaller average size than Wnt8a-HA-eGFP puncta (Figure 2K). Moreover, Wnt11b-HA-eGFP puncta have a reduced circularity compared to Wnt8a-HA-eGFP puncta (Figure 2L). Circularity is a measure of how closely an object resembles a perfect circle, with 1 representing a perfect circle and 0.1 an elongated non-circular shape. This approach can be used to test any candidate regulator of Wnt ligand diffusion. To do this, control cells should be injected with a control mRNA coding for an inactive form of the candidate regulator or an irrelevant protein such as beta-galactosidase (lacZ); this provides a more valid control than simply not injecting the regulator. Data in these examples was analysed using the non-parametric test Mann-Whitney U18-19. A statistical analysis programme was used to perform the test.
In Figure 3, we investigate Wnt ligand diffusion. A single blastomere was injected at the 4-cell stage such that the animal cap explants represent a field of cells in which only some cells express either Wnt8a-HA-eGFP or Wnt11b-HA-eGFP. By including a cell lineage tracer, we can identify expressing versus non-expressing cells and this allows the range of Wnt8a-HA-eGFP and Wnt11b-HA-eGFP ligand diffusion away from the source cells to be analysed (Figure 3B and 3C). Due to the curvature of the animal cap explants, the maximum distance that could be reliably measured using this assay was 160µm, which was not enough to measure the absolute distance of ligand diffusion. However, by using a combination of confocal analysis, image analysis and scientific analysis and graphing software the overall shape of the Wnt-HA-eGFP morphogen gradient could be analysed (Figure 3D). This data can be used to investigate whether overexpressing another protein together with the tagged ligands in the same cells can affect Wnt secretion or diffusion. In another type of experiment (Figure 3E-3J) the effects of a potential regulator in receiving cells can be measured by overexpressing the candidate protein in clones of cells adjacent to cells expressing Wnt8a or Wnt11b-HA-eGFP. This allows any non-cell-autonomous effects of the regulator on Wnt-HA-eGFP ligand diffusion to be examined. Consistent with Figure 2, more Wnt8a-HA-eGFP can be detected diffusing away from cells than Wnt11b-HA-eGFP in both diffusion experiments.
The types of experiments described in this paper have been used to analyse the effect of Sulf1 on Shh and Wnt signalling16,17.
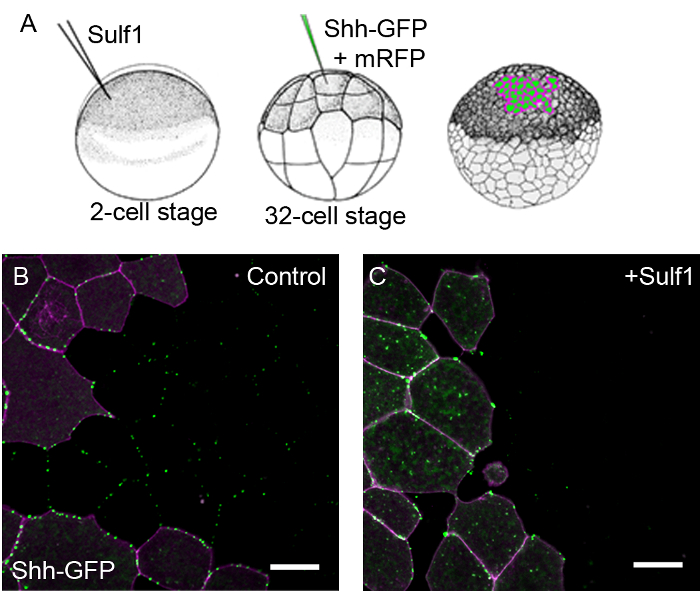
Figure 1. Modulation of Shh-GFP distribution can be detected by confocal analysis of Xenopus animal caps. (A) A cartoon depicting experimental assay where embryos are first injected with mRNA coding for Sulf1 or a control mRNA, the same embryos are later injected at the 32-cell stage with mRNA coding for Shh-GFP into a single cell. (B-C) Animal cap explants were taken at stage 8 and imaged after 4 hr. Images are shown at the edge of a Shh-GFP + memRFP expressing clone of cells. In control embryos (B), Shh-GFP is distributed away from the memRFP marked clone of signal producing cells. In embryos expressing Sulf1 (C), Shh-GFP is more restricted in its distribution, see16. memRFP is shown in magenta and Shh-GFP in green. Scale bars represent 20 µm. Please click here to view a larger version of this figure.
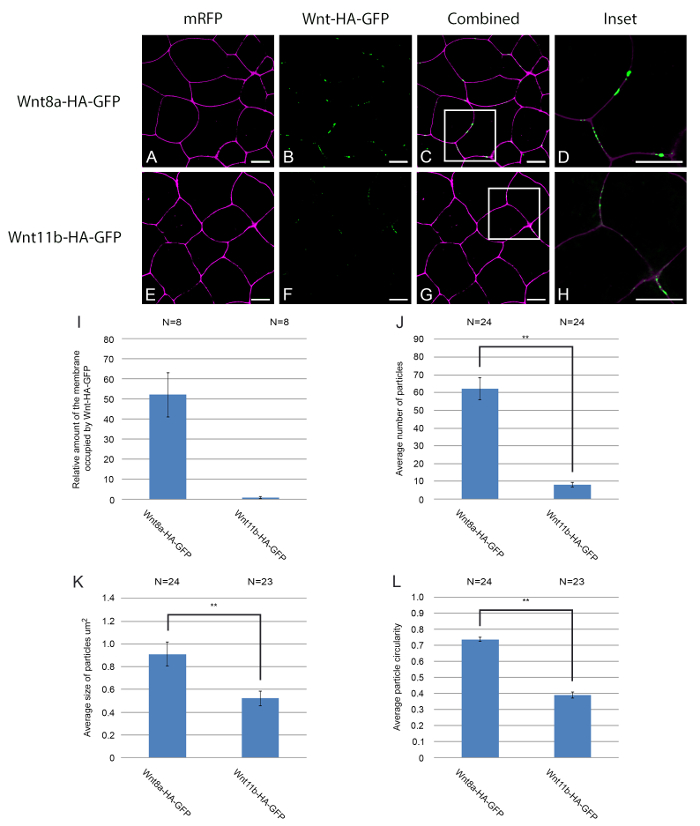
Figure 2. Quantitative and qualitative analysis of Wnt8a and Wnt11b-HA-eGFP puncta on the cell membrane. (A-H) Embryos were microinjected bi-laterally with mRNA encoding memRFP (500 pg) into the animal hemisphere at the two cell stage. In addition embryos were injected with mRNA encoding (A-D) Wnt8a-HA-eGFP (500 pg) or [E-H] Wnt11b-HA-eGFP (1ng). The embryos were also injected with mRNA coding for LacZ to provide a control for additional mRNA/protein when analysing the effects of any potential regulator. The white boxes in (C) and (G) mark the areas enlarged in panels (D) and (H) respectively. The total amount Wnt-HA-eGFP fluorescence co-localising with the cell membrane was calculated using image and script analysis software and then normalised (I). Qualitative information was extracted using image analysis software. Wnt8a and Wnt11b-HA-eGFP punctae were analysed for particle number (J), particle size (K) and particle circularity (L). Mann-Whitney U (**P<0.01), N=number of embryos. memRFP is shown in magenta, and Wnt8a/11b-HA-GFP is shown in green, scale bars represent 20µm. Please click here to view a larger version of this figure.
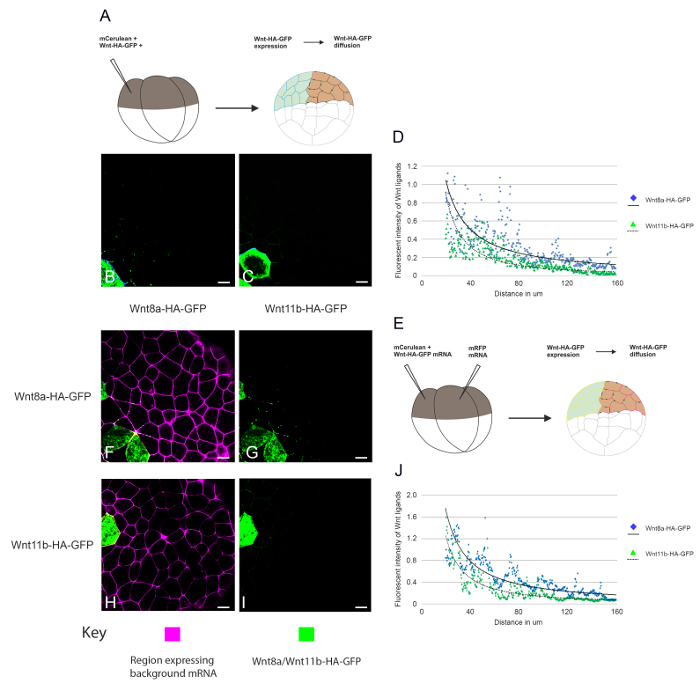
Figure 3. Measuring the range of diffusion of fluorescently tagged Wnt ligands. (A) Diagram depicting the assay used to measure Wnt8a and Wnt11b-HA-eGFP secretion and diffusion away from expressing cells. (B-C) mRNA encoding (B) memCerulean (600 pg), LacZ (4ng) and Wnt8a-HA-eGFP (2ng) or (C) memCerulean (600 pg), LacZ (4ng) and Wnt11b-HA-eGFP (2 ng) was injected into the animal hemisphere of one blastomere at the four cell stage. (D) The range of diffusion of Wnt8a-HA-eGFP and Wnt11b-HA-eGFP was measured through a control background. (E) Diagram depicting the assay used to measure Wnt8a and Wnt11b-HA-eGFP diffusion through a background expressing LacZ, see method for details. (F-G) mRNA encoding memCerulean (600 pg) and (F and H) Wnt8a-HA-eGFP (2 ng) or (G and I) Wnt11b-HA-eGFP was injected into the animal hemisphere of one blastomere at the four cell stage. An adjacent blastomere was injected with mRNA encoding memRFP (600 pg) and LacZ (4 ng) see Key for details. (J) The range of Wnt8a-HA-eGFP and Wnt11b-HA-eGFP diffusion was measured through a background expressing LacZ. The data was quantified and plotted using confocal analysis, image analysis and scientific analysis and graphing software. memCerulean (blue (C-D) and Yellow (F and H)), Wnt-HA-eGFP (green), memRFP (magenta), scale bars represent 20 µm. Please click here to download a larger version of this file.

Table 1. Primers used to subclone Wnt8a/Wnt11b-HA-eGFP and Shh-GFP into pCS2.
| Reaction | Components | |
| Example CS2+ digest | 1.5 μg of pCS2+ | |
| 2 μl of Restriction enzyme 1 | ||
| 2 μl of Restriction enzyme 2 | ||
| 5 μl of Restriction enzyme buffer (10X) | ||
| Made up to 50 μl with molecular grade water | ||
| Example mRNA synthesis | 2 μl linearised template | |
| 2 μl 10x Megascript trx mix | ||
| 2 μl 50mM ATP | ||
| 2 μl 50mM CTP | ||
| 2 μl 50mM UTP | ||
| 2 μl 5mM GTP | ||
| 2.5 μl 40mM Cap Analog (m7G(5') | ||
| 2 μl SP6 enzyme mix | ||
| 3.5 μl Molecular grade water | ||
| Example PCR reaction | 0.5 μl High fidelity DNA polymerase (2,000 units/ml) | |
| 2 ng Template DNA | ||
| 2.5 μl of forward and reverse primers (10μM) | ||
| 0.5 μl of dNTPs | ||
| 5 μl of DNA ploymerase buffer (10X) | ||
| Made up to 50 μl with molecular grade water | ||
| Example PCR conditions | Intial denaturation 2 min at 98 °C | |
| 15 sec 98 °C | ||
| 15 sec 65 °C | 30 cycles | |
| 40 sec 72 °C | ||
| Final extension 10 min at 72 °C | ||
| Example PCR product digest | 28 μl of PCR product | |
| 2 μl of Restriction enzyme 1 | ||
| 2 μl of Restriction enzyme 2 | ||
| 5 μl of Restriction enzyme buffer (10X) | ||
| 13 μl Molecular grade water | ||
| Example T4 ligation | 1 μl Cut CS2+ | |
| 3 μl Cut PCR product 1 | ||
| 3 μl Cut PCR product 2 | ||
| 1 μl of T4 DNA ligase | ||
| 1 μl T4 DNA ligase buffer (10X) | ||
| 1 μl Molecular grade water | ||
| Example template digest | 5 ug Plasmid DNA | |
| 10 μl Restriction enzyme buffer (10X) | ||
| 3 μl Not1 (except Shh-GFP, Kpn1 is used) | ||
| Made up to 100 μl with molecular grade water |
Table 2. Example reaction conditions used for subcloning and producing synthetic mRNA for Wnt8a-HA-eGFP.
| Solution | Components |
| Cysteine-HCL | 0.1X NAM |
| 2.5% L-cysteine hydrochloride monohydrate (pH7.8) | |
| NAM salts | 110 mM NaCL |
| 2 mM KCl | |
| 1 mM CA(NO3)2 | |
| 0.1 mM EDTA | |
| NAM/2 | 0.5X NAM salts |
| 5 mM HEPES pH7.4 | |
| 0.25 mM Bicarbonate | |
| 25 ug/ml Gentamycin | |
| NAM/3 + Ficoll | 0.33X NAM salts |
| 5 mM HEPES pH7.4 | |
| 0.25 mM Bicarbonate | |
| 2 5ug/ml Gentamycin | |
| 5 % Ficoll | |
| NAM/10 | 0.1X NAM salts |
| 5 mM HEPES pH7.4 | |
| 25 ug/ml Gentamycin |
Table 3. Solutions used during the production and microinjection of Xenopus laevis embryos.
