In neuroHuMiX, we co-cultured three different cell types together-bacterial, epithelial, and neuronal cells (Figure 1). To make sure the cells were all viable, we performed different assays on the different cell types. For example, we performed CFU counts on bacterial cells, cell count and cell viability assays on the epithelial cells, while the neuronal cells were assessed via microscopic analyses.
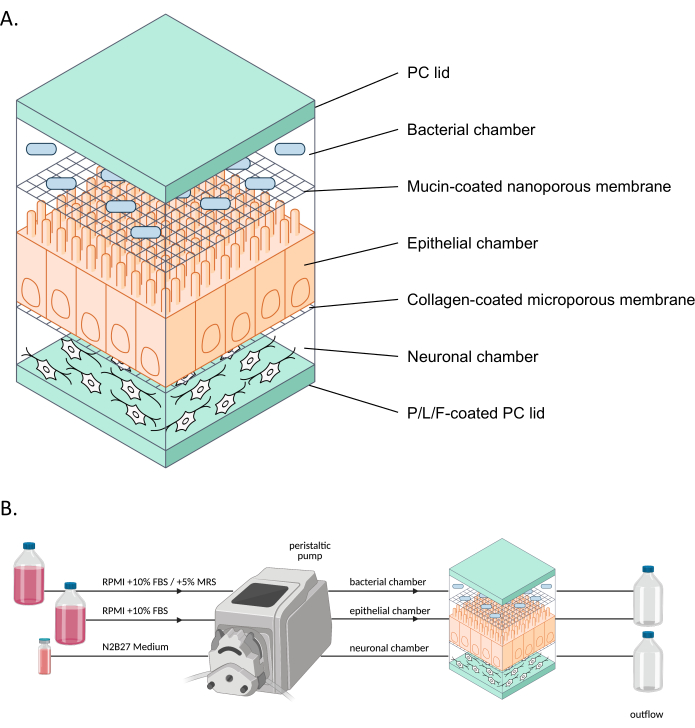
Figure 1: Schematic representation of neuroHuMiX and its experimental setup. (A) The three chambers are held between two PC lids to keep them closed. Each chamber is filled with a specific medium for the cells grown inside. The different chambers are separated by semi-permeable membranes allowing cell communication via soluble factors passing the membranes. (B) Representation of the neuroHuMiX setup. Each chamber is connected to different media bottles. For the bacterial chamber, for the first 12.5 days, the chamber is connected to RPMI + 10% FBS, before being changed for the last 36 h to RPMI + 10% FBS + 5% MRS. Abbreviations: PC = polycarbonate; P/L/F = poly L-ornithine/laminin/fibronectin; RPMI = Roswell Park Memorial Institute cell culture medium; MRS = De Man, Rogosa, and Shapre culture medium. Please click here to view a larger version of this figure.
To determine whether the cells were appropriately attached, upon opening the devices, we assessed the formation of a cell layer on the collagen-coated membrane (Figure 6A). To make sure the cells in the device were viable, an automated cell counter count (Figure 6B) and a trypan blue exclusion assay cell count were performed (Figure 6C). The assays were performed on Caco-2 cells from three different HuMiX setups: (i) Caco-2 in culture with ENs, (ii) Caco-2 in culture with L. reuteri, and (iii) the device involving co-culture of all three cell types. Statistical testing using a one-way ANOVA did not yield any significant differences between the cell types, suggesting that the Caco-2 cells remained viable in all these initial device setups and conditions tested in this study. This underlines the fact that the bacterial density reached during the co-culture of L. reuteri and the two human cell types do not have cytotoxic effects on the human cells.
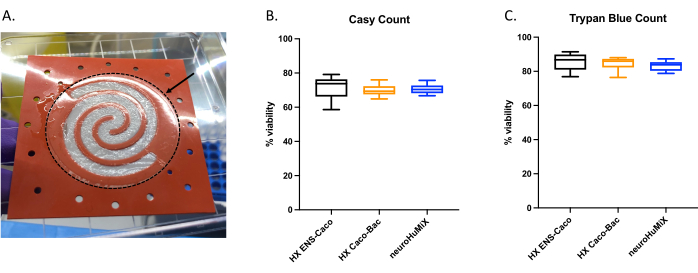
Figure 6: Assessment of Caco-2 cells on the collagen-coated membrane. (A) Layer of Caco-2 cells on the collagen-coated membrane after opening. The arrow indicates the collagen-coated membrane, which is surrounded by a dashed circle. The Caco-2 cells were growing on the spiral shape on the membrane. Cell viability of Caco-2 cells after 14 days in HuMiX. Cell counts were obtained using (B) the automated cell counter and (C) the trypan blue exclusion assay cell count. Caco-2 cell counts were determined from different culture setups in the initial device: co-culture with enteric neurons (ENs) (black), co-culture with L. reuteri (orange), and in the device (ENs and L. reuteri) (blue). A one-way ANOVA was performed, showing there is no significant difference between the different culture setups (one-way ANOVA, p = 0.1234 [ns]; error bars indicate standard error). Please click here to view a larger version of this figure.
To be able to culture L. reuteri with mammalian cells, we first optimized and adapted the culture media for use in the device. We found that a 5% mix of MRS in RPMI 1640 (supplemented with 10% FBS) was optimally suited for the growth of L. reuteri, while not being cytotoxic for the mammalian cells used in these assays. Subsequently, a CFU count was performed to assess the growth of L. reuteri when cultured in the device for 24 h. The CFU count was assessed for two different initial device setups (Figure 7)-L. reuteri co-cultured with Caco-2 and L. reuteri in the device. In both setups, the CFU counts were significantly different from the HuMiX inoculum and the harvested cells (one-way ANOVA, p = 0.0002), indicating growth of the bacterial cells inside the initial device.
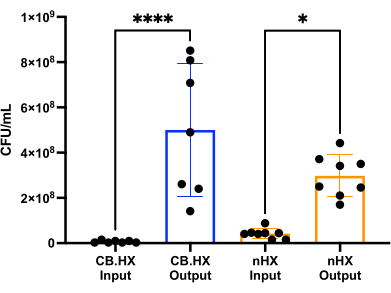
Figure 7: Limosilactobacillus reuteri CFU count of the inoculum (diluted 1:100,000) and after 24 h in HuMiX. Two different setups: Caco-2 cells in co-culture with L. reuteri and the device. A one-way ANOVA shows a significant difference (p = 0.0002 [***]) between the inoculum and the harvested cells, meaning the bacteria are growing inside HuMiX. Error bars indicate the standard error. Abbreviations: CB.HX = Caco-2 bacteria HuMiX; nHX = neuroHuMiX. Please click here to view a larger version of this figure.
To assess whether culturing the ENs within the device would alter the phenotype of the cells, the gross morphology of the ENs was observed using an inverted phase-contrast microscope. During this step, both the confluency and EN morphology were assessed. Establishment of a confluent neuronal network indicated that the cells had attached well onto the coated device's PC lid. Importantly, this highlights the notion that they grew in co-culture with Caco-2 and L. reuteri. The edge between the confluent neuronal network and the gasket-delineated spiral was clearly apparent (Figure 8).
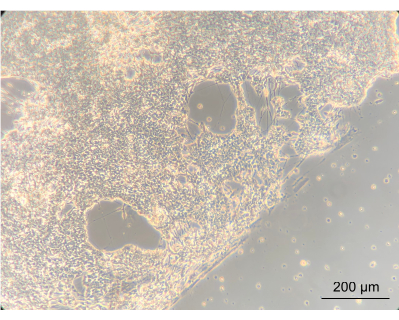
Figure 8: Enteric neurons after 14 days of culture in the device. On the left side of the image, the neurons have grown to a confluent layer on the spiral. The edge, between the neuronal layer and the space without cells, is the edge of the spiral; magnified 10x, scale bar = 200 µm. Please click here to view a larger version of this figure.

Figure 2: Lids used in the device. Images show top (left) and bottom (right) PC lids. Each side of the PC lid is 6.4 cm. Abbreviation: PC = polycarbonate. Please click here to view a larger version of this figure.

Figure 3: Epithelial chamber gasket on bottom PC lid. Top view of the epithelial chamber gasket placed on the bottom PC lid (left), and bottom view (right) showing the alignment of the epithelial chamber gasket with the inlets and outlets of the bottom PC lid. Each side of the gaskets, as well as the PC lid, measures 6.4 cm. Abbreviation: PC = polycarbonate. Please click here to view a larger version of this figure.

Figure 4: Assembly of the device. (A) Different parts for assembling HuMiX: (1) bottom PC lid; (2) gasket with collagen-coated microporous membrane, which is placed on top of (1); (3) sandwich gasket with a mucin-coated nanoporous membrane in between and placed on top of (2); (4) top PC lid placed on top of (3). Each side of the gaskets and PC lids measures 6.4 cm. (B) All parts from (A) placed together. (C,D) Assembled device-top (left) and side (right) view. B is placed into the clamping system to close the system. (C) Each side of the top clamp measures 8 cm. Abbreviation: PC = polycarbonate. Please click here to view a larger version of this figure.
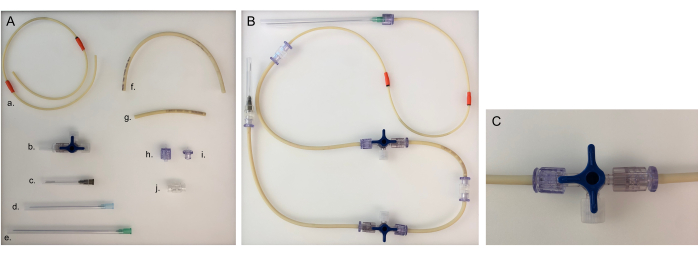
Figure 5: Parts needed for tubing line and assembled tubing line for one chamber. (A) Different parts to build a tubing line: a. pump tubing line; b. three-way stopcock; c. 40 mm needle; d. 80 mm needle; e. 120 mm needle; f. long tubing line (20 cm); g. short tubing line (8 cm); h. male Luer; i. female Luer; j. adaptor. (B) Assembled tubing line for the bacterial or epithelial chamber. For the neuronal chamber, the 120 mm needle would need to be changed to an 80 mm needle. (C) Three-way stopcock valve turned to redirect the medium flow from the device to the 'open connector' and to close the chamber. Please click here to view a larger version of this figure.
| Day | 0 | 2 | 4 | 6 | 8 | 10 |
| Media Composition | 100% E6 | 100% E6 | 75% E6 | 50% E6 | 25% E6 | 100% N2 |
| + LDN | + LDN | 25% N2 | 50% N2 | 75% N2 | + LDN | |
| + SB | + SB | + LDN | + LDN | + LDN | + SB | |
| + CHIR | + SB | + SB | + SB | + CHIR | ||
| + CHIR | + CHIR | + CHIR | + RA | |||
| + RA | + RA | |||||
| Molecule | [concentration] | |||||
| LDN | 100 nM | |||||
| SB | 10 µM | |||||
| CHIR | 3 µM | |||||
| Retinoic Acid (RA) | 1 µM |
Table 1: Media composition.
| Media | Components (concentrations listed in Table of materials) | Volume (mL) |
| N2 media (50 mL) | DMEM-F12 | 48 |
| N2 Supplement | 0.5 | |
| L-Glutamine | 0.5 | |
| Penicillin/Streptomycin | 0.5 | |
| NEAA | 0.5 | |
| N2B27/ENS Media (50 mL) | Neurobasal | 48 |
| N2 Supplement | 0.5 | |
| L-Glutamine | 0.5 | |
| Penicillin/Streptomycin | 0.5 | |
| B27-A | 0.5 |
Table 2: Media recipes.
| Sterilization Temperature (°C) | 116 |
| Sterilization Time (min) | 20 |
| Dry Time (min) | 10 |
| Pulses | 3 |
| End Temperature (°C) | 99 |
Table 3: HuMiX autoclave run.
| Rotations per minute (rpm) | Average flow rate (µL/min) |
| 0.5 | 13 |
| 2 | 79 |
| 5 | 180 |
Table 4: Flow rates of the peristaltic pump.
