Isolation and culture of UCA-PSCs, UCV-PSCs, and WJ-MSCs from the umbilical cord
The umbilical arteries, umbilical vein, and Wharton's jelly were mechanically separated from the umbilical cord and cut into 2-3 cm3 pieces. The distance between arteries, vein, or Wharton's jelly tissue blocks was approximately 1 cm, arranged in a quincunx shape (Figure 1A–C). The three kinds of stem cells were isolated by a tissue-attached culture method5. After approximately 1 week of attached culture, the cells were observed to crawl out radially, exhibiting a long spindle shape and rapid proliferation (Figure 1D–F). The cells reached 70%-80% confluence at approximately 2 weeks, and the growth rate was accelerated after passage. The cells either grew in an even spindle, parallel arrangement, or vortex growth (Figure 1G–I). In addition, the different stem cells had similar proliferation tendencies as demonstrated by the CCK-8 assay (Supplementary Figure 1). However, on days 4 and 5, UCA-PSCs had a significant higher growth rate compared to UCV-PSCs (P < 0.05).
UCA-PSCs, UCV-PSCs, and WJ-MSCs expressed MSC surface markers
The surface markers of third-generation stem cells were identified by flow cytometry. As shown in Figure 2A–O, UCA-PSCs, UCV-PSCs, and WJ-MSCs all highly expressed the MSC-specific surface markers CD13 and CD73, but did not express CD34 and CD45, which are endothelial cell markers and hematopoietic stem cell markers. The cells did not express HLA-DR, a hematopoietic stem cell marker2. The characterization of Adipose-derived stem cells (ADSCs) as a control was displayed in Supplementary Figure 2A-E, which indicates that the stem cells expressed the surface marker of MSCs consistent with that of ADSCs. MSCs were incubated with FITC-IgG and were negative for the FITC signal (Supplementary Figure 3A). In addition, the results of three repeated experiments were analyzed in Supplementary Figure 4, which proved the method was reproducible.
UCA-PSCs, UCV-PSCs, and WJ-MSCs possessed adipogenic differentiation potential
After adipogenic induction, the morphology of the isolated stem cells changed from long spindle to round. After approximately 12 days of adipogenic induction, round lipid droplets were observed in the cells, which had strong refraction. Some lipid droplets fused to form large lipid droplets. On approximately the 14th day, the three kinds of cells showed red lipid droplets of different sizes by oil red O staining (Figure 3A-C). The characterization of ADSCs as a control was displayed in Supplementary Figure 2F, which indicates that the different stem cells possessed adipogenic differentiation potential consistent with that of ADSCs. For adipogenic differentiation, there were no lipid droplets in the human endometrial stromal cells (ESCs) (Supplementary Figure 3B).
UCA-PSCs, UCV-PSCs, and WJ-MSCs have osteogenic differentiation potential
After osteogenic induction, the cell morphology changed from a long spindle to a lamellar structure, and the extracellular matrix began to deposit. After osteogenic induction for 10 days, calcium nodules appeared in the three kinds of cells. At approximately 21 days after induction, the cell morphology was polygonal, and a calcium nodule-like structure was seen in the center of the cells. The cells showed calcified nodules in the middle, as shown by Alizarin red staining (Figure 4A-C). The characterization of ADSCs as a control was displayed in Supplementary Figure 2G, which indicates that stem cells possessed osteogenic differentiation potential consistent with that of ADSCs. For osteogenic differentiation potential, there were no calcified nodules in the ESCs (Supplementary Figure 3C).
UCA-PSCs, UCV-PSCs, and WJ-MSCs have neurogenic differentiation potential
The three kinds of stem cells were induced by neurogenic induction solution; the spindle-shaped fibroblast-like cells became floated ball-shaped neurosphere-like cells. The cellular synapses were formed in all the cell variants. The morphology of the cells gradually became stellate and interconnected, and had nerve-like characteristics and enhanced refraction. The results of cellular immunofluorescence showed that the stem cells expressed neuron-specific markers of NSE after induction (Figure 5A-C). The results showed that the cells had multidirectional differentiation potential after induction. The characterization of ADSCs as a control was displayed in Supplementary Figure 2H, which indicates that the stem cells possessed neurogenic differentiation potential consistent with that of ADSCs. For neurogenic differentiation potential, there was no NSE signal in the ESCs (Supplementary Figure 3D).
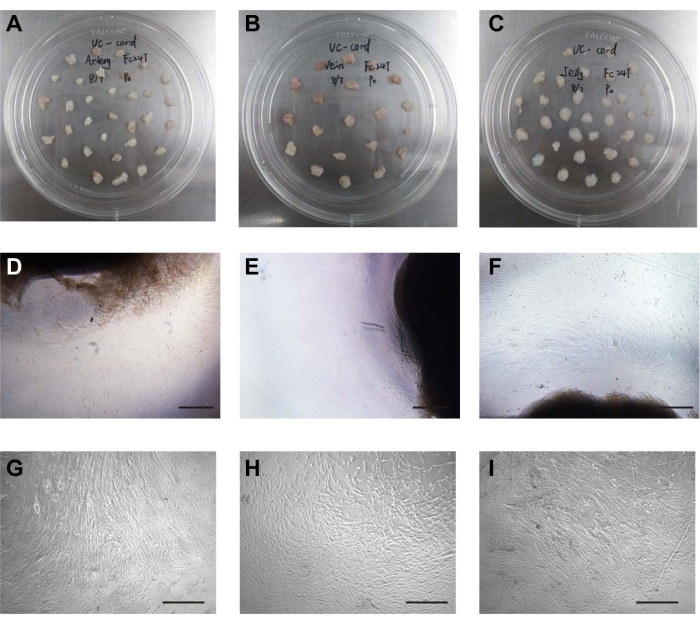
Figure 1: Isolation and culture of cells. (A) The umbilical arteries were placed in the cell culture dish. (B) The umbilical vein was placed in the cell culture dish. (C) Wharton's jelly was placed in the cell culture dish. (D) The stem cells crawled out of the umbilical artery tissue. Scale bar = 500 µm. (E) The stem cells crawled out of umbilical vein tissue. Scale bar = 500 µm. (F) The stem cells crawled out of the Wharton's jelly tissue. Scale bar = 500 µm. (G) Morphology of cultured UCA stem cells after passage. Scale bar = 500 µm. (H) Morphology of cultured UCV stem cells after passage. Scale bar = 500 µm. (I) Morphology of cultured WJ stem cells after passage. Scale bar = 500 µm. Please click here to view a larger version of this figure.
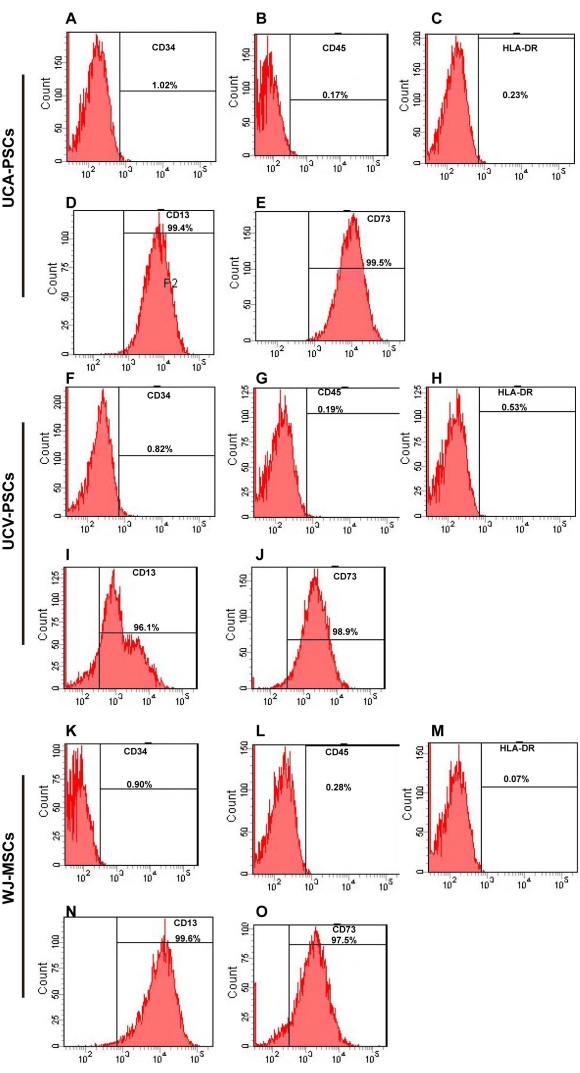
Figure 2: Flow cytometry analysis of UCA-PSC, UCV-PSC, and WJ-MSC surface markers. (A-E) UCA-PSCs were negative for CD34, CD45, and HLA-DR. UCA-PSCs were positive for CD13 and CD73. (F–J) UCV-PSCs were negative for CD34, CD45, and HLA-DR. UCV-PSCs were positive for CD13 and CD73. (K–O) WJ-MSCs were negative for CD34, CD45, and HLA-DR. WJ-MSCs were positive for CD13 and CD73. The x-axis is the amount of fluorescence. Please click here to view a larger version of this figure.
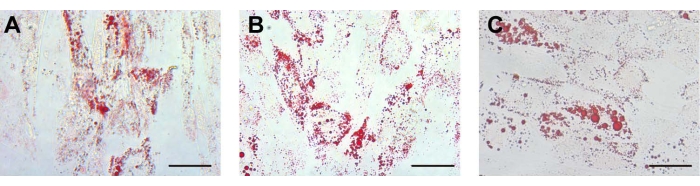
Figure 3: Adipogenic differentiation. For adipogenic differentiation, the lipid droplets in the (A) UCA-PSCs, (B) UCV-PSCs, and (C) WJ-MSCs cultured in an adipogenic induction medium were stained with oil red O. Scale bar, 50 µm. Please click here to view a larger version of this figure.
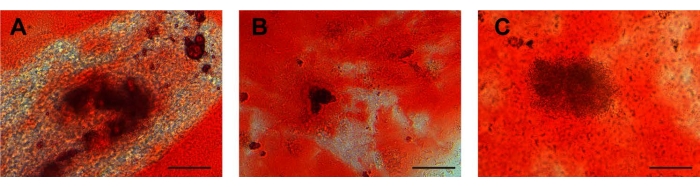
Figure 4: Osteogenic differentiation. Calcium deposits in (A) UCA-PSCs, (B) UCV-PSCs, and (C) WJ-MSCs cultured in an osteogenic induction medium were stained with Alizarin red for osteogenic differentiation. Purple-red clumps are calcium nodules. Scale bar = 50 µm. Please click here to view a larger version of this figure.
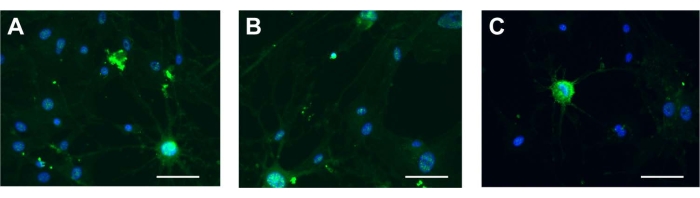
Figure 5: Neurogenic differentiation. NSE in (A) UCA-PSCs, (B) UCV-PSCs, and (C) WJ-MSCs cultured in a neurogenic induction medium. Green shows NSE stain, and blue shows nucleus. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Supplementary Figure 1: Cell proliferation. CCK-8 assays reflected that the different stem cells had similar proliferation tendencies. On days 4 and 5, UCA-PSCs had a significant higher growth rate than UCV-PSCs. The error bars represent the means ± SE of the three independent experiments performed in triplicate. P < 0.05. Please click here to download this File.
Supplementary Figure 2: Characterization of Adipose-derived stem cells (ADSCs) as the positive control. (A–E) ADSCs were negative for CD34, CD45, and HLA-DR. ADSCs were positive for CD13 and CD73. (F) For adipogenic differentiation, the lipid droplets in ADSCs cultured in an adipogenic induction medium were stained with oil red O. Scale bar = 50 µm. (G) Calcium deposits in ADSCs cultured in an osteogenic induction medium were stained with Alizarin red for osteogenic differentiation. Scale bar = 50 µm. (H) NSE in ADSCs cultured in neurogenic induction medium. Scale bar = 50 µm. Please click here to download this File.
Supplementary Figure 3: Characterization of MSCs and human endometrial stromal cells (ESCs) as the negative control. (A) MSCs were incubated with FITC-IgG and were negative for the FITC signal. (B) For adipogenic differentiation, there were no lipid droplets in ESCs. Scale bar = 50 µm. (C) For osteogenic differentiation, there were no calcium deposits in ESCs. Scale bar = 50 µm. (D) There was no NSE signal in ESCs. Scale bar = 50 µm. Please click here to download this File.
Supplementary Figure 4: Profiles of cell surface markers in the stem cells, and IgG. UCA-PSCs, UCV-PSCs, and WJ-MSCs were negative for CD34, CD45, and HLA-DR. UCA-PSCs, UCV-PSCs, and WJ-MSCs were positive for CD13 and CD73. IgG as overlay isotype control was negative for CD34, CD45, HLA-DR, CD13, and CD73. The results of three repeated experiments were analyzed, which proved the method was reproducible. The error bars represent the means ± SE of three independent experiments performed in triplicate. Please click here to download this File.
