Quantifying the percentage of synaptic spinules within the excitatory presynaptic bouton population in ferret primary visual cortex
Although spinule-like protrusions from neurites into excitatory presynaptic boutons have been observed for decades19,26, their potential importance for synaptic function has remained obscure. These experiments were designed to determine the proportion of excitatory presynaptic boutons containing spinules throughout postnatal development in the ferret primary visual cortex (V1) to ascertain the potential importance of spinules for synaptic function in relation to developmental milestones. Accordingly, aligned 4 nm/voxel isotropic FIB-SEM image volumes were acquired from a postnatal day (p)21 (15.1 x 14.1x 2.8 µm), p46 (9.7 x 8.4 x 2.7 µm), p60 (24.2 x 16.2 x 2.4 µm), and >p90 (24.2 x 16.2 x 2.4 µm) ferret V1, imaged using an FEI Helios 660 DualBeam FIB-SEM at 52° tilt, 4.2 mm working distance, 3 kV acceleration voltage, and 400 pA current in backscatter mode. Although stacks were roughly aligned with FEI software on acquisition, all stacks went through subpixel alignment using Fiji (refer to 1.4.3). These ages correspond to before the onset of correlated visual experience (i.e., eye-opening; p21), at the height of the canonical critical period for ocular dominance plasticity in ferret V1 (p46), near the end of the critical period (p60), and late adolescence (>p90).
Each image volume was scaled using Fiji, and every excitatory presynaptic bouton within the four FIB-SEM volumes was identified. Excitatory synapses were identified by the presence of parallel presynaptic and postsynaptic membranes, a prominent asymmetric (Gray's Type I) postsynaptic density27, and ≥3 presynaptic vesicles. Every excitatory presynaptic bouton was evaluated across its 3D volume for the presence of one or more spinules based on conservative criteria (refer to 4.5) and to determine whether the postsynaptic density (PSD) had a discontinuity termed a perforation that is associated with higher rates of plasticity28,29. In addition, each spinule was followed back to its parent structure to determine the proportion of spinules that emanate from distinct neurites or glia. Toward this end, regions of interest (ROIs) were outlined using the oval tool in Fiji for each excitatory presynaptic bouton by encircling the entire synapse (see 2.2.1 – 2.2.3). Each ROI contained the number of the boutons (in sequential order) and noted the presence/absence of a spinule, the postsynaptic target (e.g., dendritic spine or dendritic shaft), and the presence/absence of a perforated PSD (see Note after 2.2.3). By examining every excitatory bouton within these four image volumes, the percentages of presynaptic boutons that contained spinules were determined to increase across development. Yet, the 3D relationships of spinules to presynaptic boutons remained to be determined.
Examining the relationship between synaptic spinules and their encapsulating excitatory presynaptic boutons
To examine the relationship between the two most abundant spinule types (i.e., spinules emanating from postsynaptic spines and those projecting from adjacent axons) and their encapsulating excitatory spinule-bearing presynaptic boutons (SBBs), the >p90 image volume was analyzed to determine the sizes of these spinule types and whether these spinules were engulfed by similar-sized SBBs. Using the ROIs from the spinule prevalence quantification in Fiji described above, SBBs that contained spinules from postsynaptic spines or adjacent axons/boutons were identified. After examining these SBBs and spinules in 3D within Fiji, it was determined that a section thickness of 8 nm was sufficient to resolve the thin invagination of each spinule into its respective SBB. As such, substacks containing every other section (i.e., 8 nm z resolution) were made, with a substack range that included the full 3D extent of SBBs and their spinules (see 3.3). After transferring these substacks to Reconstruct, 11 SBBs containing postsynaptic spine spinules, and 14 SBBs containing adjacent axons spinules were traced and three-dimensionally reconstructed (see 4.4-4.7). These analyses revealed that postsynaptic spine spinules were 2.7 times larger than adjacent axon spinules (0.016 ± 0.005 µm3 vs. 0.0059 ± 0.001 µm3, mean ± SEM, postsynaptic spine vs. adjacent axon spinules, respectively; Figure 3C). However, given the small sample sizes for this pilot study, these data were not statistically significant at p < 0.05 (Mann Whitney U test, two-tailed, U = 56, p = 0.26). Using a freely available effect size and statistical power analysis software (G*Power)30, it is estimated that if this medium effect size (0.596) for the difference in spinule volumes holds, ~60 more postsynaptic spine and adjacent axon spinules reconstructions will be needed to obtain a statistically significant result with α = 0.05 and statistical power of 0.95. Interestingly, these analyses also found that while the volumes of SBBs containing postsynaptic spine spinules were similar to those of SBBs containing adjacent axon spinules (0.21 ± 0.04 µm3 vs. 0.18 ± 0.02 µm3, postsynaptic spine vs. adjacent axon containing SBBs, respectively), the volume that adjacent axon spinules occupied within their enveloping SBBs was nearly identical to the volume that postsynaptic spine spinules occupied within their SBBs (19.3 ± 3.2 % vs. 17.5 ± 2.4 %, postsynaptic spine vs. adjacent axon spinules, respectively; Mann Whitney U test, two-tailed, U = 68, p = 0.64; Figure 3D). In sum, these pilot data suggest that postsynaptic spine spinules may be larger than their adjacent axon counterparts, and that adjacent axon spinules may preferentially invaginate into a population of relatively small boutons in ferret V1.
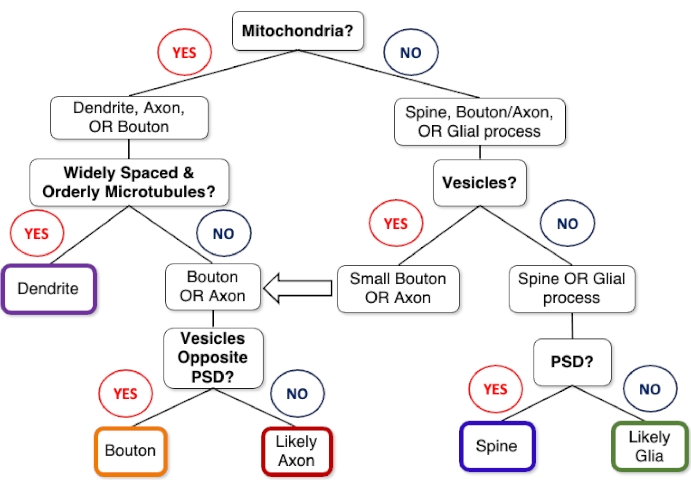
Figure 1: Decision tree for identifying neurites within FIB-SEM images. Neurite cross-sections (e.g., longitudinal and transverse sections) within FIB-SEM images can be identified based on the presence/absence of a few key organelles. For example, neurites containing mitochondria include dendrites, axons, and presynaptic boutons. Yet, under most FIB-SEM staining protocols, only dendrites will have a prominent, widely spaced (~55-70 nm spacing)31, and orderly arrangement of microtubules. In contrast, axons and boutons display neurotransmitter-containing vesicles, with boutons exhibiting a dense pool of these synaptic vesicles at their active zone(s) opposite the PSD, while axons contain dense (~13-30 nm spaced)31 microtubules and lower contrast neurofilaments. Dendritic spines nearly always lack mitochondria, microtubules, and vesicles, and therefore can most readily be differentiated from glial processes (e.g., astrocytes) based on the presence of a PSD. However, spines are also mostly larger than glial processes, connect to their parent dendrite, and sometimes contain a spine apparatus. Abbreviations: FIB-SEM = focused ion beam scanning electron microscopy; PSD = postsynaptic density. Please click here to view a larger version of this figure.
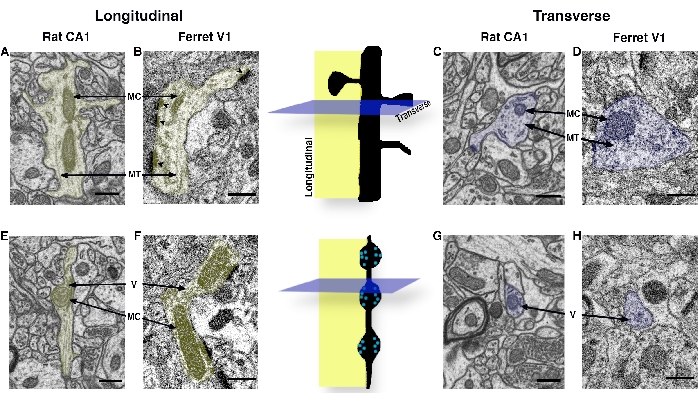
Figure 2: Neurite cross-section identification primer, displaying neuronal dendrites and axons from adult rat CA1 hippocampus and late adolescent ferret V1. As neuronal dendrites and axons have a range of sizes and course through the brain at angles tangential to the plane of sectioning, similar-sized dendrites and axons cut at different angles have unique appearances within FIB-SEM images. Central cartoons show a dendrite (above) and an axon (below) sectioned in a longitudinal plane (yellow) and a transverse plane (blue). (A, B) Neuronal dendrites sectioned in a longitudinal plane (i.e., along their long axis). Arrows point to distinguishing mitochondria (MC) and orderly widely spaced 'stripe-like' microtubules (MT). Note that dendrites followed along their depth should also exhibit PSD (arrowheads; dark, electron-dense regions along dendrite and spine in B). (C, D) Neuronal dendrites sectioned in a transverse plane (i.e., along their short axis). Arrows point to MT cut in a transverse plane that appear vesicular and MC that appear circular or ovoid. Dendrites can be differentiated from transversely-sectioned axons by their orderly, widely-spaced microtubules, ~1.25-2.75 × larger diameter32, absence of synaptic vesicles, and the presence of one or more PSDs. (E, F) Neuronal axons sectioned along a longitudinal plane, displaying MC and vesicles (V). Note that the axon in E contains orderly, densely-spaced microtubules, potentially leading to its misidentification as a dendrite, yet it also contains prominent neurotransmitter-containing vesicles. Clustered synaptic vesicles are also seen in the axon in F at two active zones opposite PSDs. Axons followed through the depth of most image volumes will display en passant synaptic boutons along their length, as in F. (G, H) Neuronal axons sectioned in a transverse plane, displaying prominent vesicles. Axons sectioned in a transverse plane often appear ovoid and have regions along their depth that are among the smallest diameter neuronal structures in the neuropil. Scale bars in A–H = 0.5 µm. Abbreviations: FIB-SEM = focused ion beam scanning electron microscopy; PSD = postsynaptic density; MC = mitochondria; MT = microtubules; V = vesicles. Please click here to view a larger version of this figure.
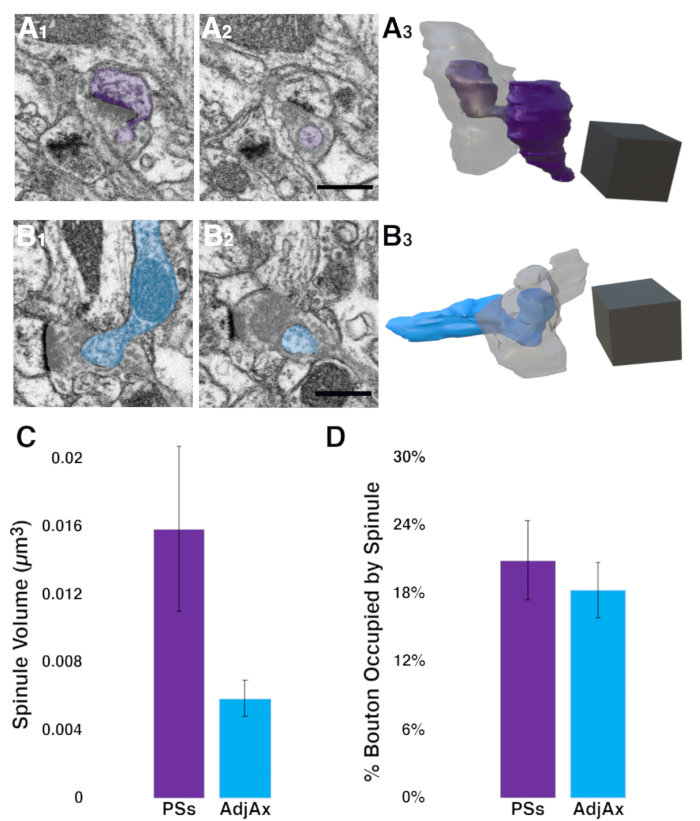
Figure 3. Pilot study using the described analysis pipeline to quantify postsynaptic spine and adjacent axon spinule volumes within presynaptic boutons in ferret V1. (A1–A3) Two sequential images showing the invagination (A1) and envelopment (A2) of a postsynaptic spine spinule (purple) into an excitatory presynaptic bouton (gray). Full 3D reconstruction (A3) showing the postsynaptic spine (purple) projecting a large anchor-like spinule into its presynaptic bouton (gray, made transparent) partner. (B1–B3) Two sequential images showing an adjacent (i.e., non-synaptic) axon (cyan) invaginating into (B1) and becoming fully encapsulated (B2) within a presynaptic bouton (gray). Note that the presynaptic bouton has a synapse with a postsynaptic spine to the left of both images, indicated by the prominent asymmetric PSD. Full 3D reconstruction (B3) of this adjacent axon spinule (cyan) within a presynaptic bouton (gray, made transparent). (C) Postsynaptic spine spinules show a trend toward being larger than spinules projecting from adjacent axons (n = 11 and 14, for PSs and AdjAx spinules, respectively; error bars indicate Standard Error of the Mean (SEM); Mann Whitney U test; p =0.26). (D) AdjAx spinules occupy a similar portion of their presynaptic boutons as PSs spinules (error bars indicate SEM; Mann Whitney U test, p = 0.64.). Scale bars for A2 and B2 = 0.5 µm; scale cubes for A3 and B3 = 0.5 µm/side. Abbreviations: PSD = postsynaptic density; PSs = postsynaptic spine spinules (PSs); AdjAx = adjacent axons. Please click here to view a larger version of this figure.
| Software | OS Compatibility | Semi-automated Segmentation | User-friendly UX | 3D Measurement Features | Download Site | |
| Espina | Windows; Linux | +++ | +++ | ++++ | https://cajalbbp.es/espina/#started | |
| IMOD | Windows; Linux; Mac | + | + | ++++ | https://bio3d.colorado.edu/imod/ | |
| Neuromorph (Blender plugin) | Windows; Linux; Mac | + | ++ | +++ | https://neuromorph.epfl.ch/software/ | |
| Reconstruct | Windows | + | ++++ | ++ | https://synapseweb.clm.utexas.edu/software-0 | |
| TrakEM2 (ImageJ Plugin) | Windows; Linux; Mac | ++ | +++ | +++ | https://www.ini.uzh.ch/~acardona/trakem2.html | |
Table 1: 3D Image volume analysis software. Selection of free, open-source software programs for registration, visualization, reconstruction, and measurement of thin subcellular structures within FIB-SEM image volumes. A qualitative assessment is presented for the degree to which each software platform contains three features salient for users (semi-automated segmentation, user-friendly user interface (UX), and 3D measurement features), with "++++" as the highest degree/amount, and "+" as the lowest degree/amount. Abbreviation: FIB-SEM = focused ion beam scanning electron microscopy.
