TGF-β2 induces EndMT and stimulates Snail expression in MS-1 endothelial cells
TGF-β is one of the cytokines with greatest potential to induce EndMT. After treating MS-1 cells with TGF-β2 (1 ng/mL) for 3 days, endothelial MS-1 cells lose their cobblestone-like structure and differentiate into spindle-shaped mesenchymal-like cells (Figure 1A)15. To further verify the role of TGF-β2 in inducing cell phenotypic changes, we pre-treated the cells with the small molecule activin receptor-like kinase (ALK)4/ALK5/ALK7 inhibitor SB431542 before TGF-β2 stimulation19. SB431542 completely abrogated TGF-β2-induced cell morphology changes (Figure 1A). The TGF-β2 induced EndMT process was further investigated by studying changes in the expression of EndMT-related markers. As shown in Figure 1B, the endothelial protein Pecam-1 was potently decreased after TGF-β2 stimulation, while the mesenchymal factor Sm22α was profoundly upregulated by TGF-β215. These data are consistent with the notion that TGF-β2 triggered EndMT in MS-1 cells. Next, we investigated the effects of TGF-β2 on Snail and Slug expression. As shown in Figure 1C, Snail was markedly upregulated by TGF-β2, while Slug expression was not influenced by TGF-β2 in MS-1 cells15. The quantification of Snail expression from three independent experiments is shown in Figure 1D.
Depletion of Snail by CRISPR/Cas9 in MS-1 endothelial cells
As Snail was induced by TGF-β2 and likely involved in TGF-β2-mediated EndMT, we performed CRISPR/Cas9 gene editing to genetically deplete Snail expression in MS-1 cells. We hypothesized that the depletion of Snail would be sufficient to inhibit TGF-β2-induced EndMT. As shown in Figure 2A, we generated Snail knockout cells in two steps. Firstly, Cas9 was ectopically expressed by infecting MS-1 cells with a Cas9 expressing lentivirus. Since there is a blasticidin resistance cassette in the pLV-Cas9 construct, we checked the expression of Cas9 by Western blot analysis in blasticidin resistant cells (Figure 2D). Subsequently, we introduced sgRNAs that specifically targeted Snail to disrupt its protein expression. This procedure was also performed by infection with lentiviral particles carrying the AA19 pLKO.1-Snail-sgRNA construct, which includes a puromycin expression cassette. Cas9-expressing cells were again infected with gRNA containing lentivirus and further selected with puromycin. Two complementary sgRNA oligos targeting murine Snail were designed with a predicted low off-target activity (Figure 2B,C). After introducing two independent Snail sgRNAs in Cas9 expressing MS-1 cells, Snail protein expression was abrogated (Figure 2D)15.
Deficiency of Snail inhibits TGF-β2-induced EndMT in MS-1 cells
To demonstrate the function of Snail in TGF-β2-mediated EndMT, we performed an EndMT assay in Snail-depleted cells and compared it with parental MS-1 cells. As shown in Figure 3A, the knockout of Snail was sufficient to inhibit the fibroblast-like cell morphology driven by TGF-β2 in MS-1 cells15. In addition, the TGF-β2-mediated decline in Pecam-1 and enhancement of Sm22α were completely blocked in Snail-depleted MS-1 cells. In summary, we demonstrated that Snail is critical for TGF-β2-mediated EndMT in MS-1 cells (Figure 3B)15.
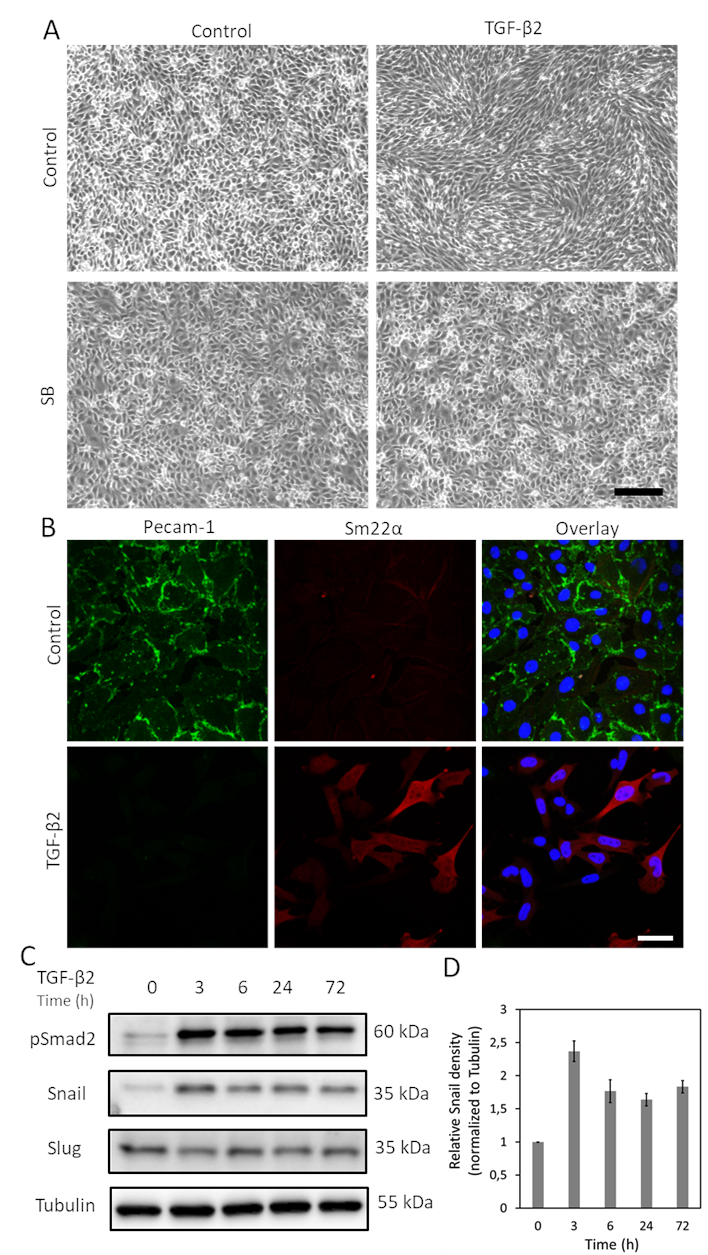
Figure 1. TGF-β2 induces EndMT and Snail expression in MS-1 cells. A. Effects of TGF-β2 and/or TGF-β type I receptor kinase inhibitor SB-431542 on cell morphology. Brightfield images of MS-1 cells upon treatment with TGF-β2 (1 ng/mL) and/or SB-431542 (SB, 5 µM, administered 30 min prior to TGF-β2) for 2 days. Scale bar represents 200 µm. B. Immunofluorescence staining of Pecam-1 (green) and Sm22α (red) in MS-1 cells cultured in medium containing TGF-β2 (1 ng/mL) for 3 days. Nuclei are visualized in blue (DAPI). Scale bar: 50 µm. C. Western blot with whole cell lysate of TGF-β2 stimulated MS-1 cells. The expression of Snail, but not Slug, was enhanced by TGF-β2 stimulation, as previously reported in Ma et al15. D. Quantification of Snail expression by integrating the results from three independent western blot experiments. Please click here to view a larger version of this figure.

Figure 2. Depletion of Snail by CRISPR-Cas9 gene editing. A. Scheme depicting how to generate Snail knockout cells. Bsd: Blasticidin. Puro: Puromycin. B. Oligonucleotides of two independent sgRNAs targeting Snail using CHOPCHOP (http://chopchop.cbu.uib.no/) and Cas-OFFinder (http://www.rgenome.net/cas-offinder/). C. The predicted off-target activity of the two gRNAs for Snail using Cas-OFFinder (http://www.rgenome.net/cas-offinder/). D. Cas9 and Snail expression in wild type (WT) and Cas9-overexpressed MS-1 measured by Western blot analysis. E. Knockout of Snail with two independent gRNAs in MS-1 cells as measured by Western blot analysis. We reported similar results in Ma et al15. Please click here to view a larger version of this figure.

Figure 3. Genetic depletion of Snail inhibits TGF-β2-induced EndMT in MS-1 cells. A. Brightfield images of MS-1 cells upon treatment with TGF-β2 (0.1 ng/mL) for 3 days in wildtype (WT, upper panel) and Snail knocked out (lower panel) cells. Scale bar represents 200 µm. B. Immunofluorescent staining for Pecam-1 (green), Sm22α (red) and nuclei (blue) of MS-1 cells cultured in medium containing TGF-β2 (1 ng/mL) for 3 days. Depletion of Snail abrogated TGF-β2-induced decrease of Pecam-1 and increase of Sm22α expression. Scale bar represents 50 µm. C. Schematic representation of the effect of Snail knockout on TGF-β-induced EndMT in MS-1 cells. TGF-β stimulates the expression of Snail through Smad pathway by phosphorylating Smad2/3 and further drives EndMT. Knocking out Snail using CRISPR/Cas9-based gene editing abrogated TGF-β-mediated EndMT. Please click here to view a larger version of this figure.
