Molecular properties and visualization of the chemical space
All compounds in the BIOFACQUIM10, PeruNPDB11, and FDA13 datasets had six physicochemical properties calculated for them. These qualities were then plotted onto violin plots, which allow one to see how the properties of the three studied datasets are distributed (Figure 1). The distribution profiles of the six physicochemical parameters of pharmaceutical interest, namely molecular weight (MW), octanol/water partition coefficient (clogP), topological surface area (TPSA), aqueous solubility (clogS), number of H-bond donor atoms (HBD), and number of H-bond acceptor atoms (HBA), differ between the datasets. However, the TPSA results demonstrated significant variances when comparing the BIOFACQUIM and FDA datasets to PeruNPDB. Using PCA, the dataset's chemical space visualization was carried out. However, 3D-visual PCA analysis reveals that molecules in both datasets of NPs roughly overlap chemical space with the FDA's collection of approved pharmaceuticals. While in some areas, chemicals from PeruNPDB or BIOFACQUIM predominate (Figure 2).
Diversity analysis
In addition, a CDP based on molecular fingerprints, scaffolds, and physicochemical attributes was utilized to assess the diversity of the datasets. The property-based diversity of the databases from PeruNPDB, BIOFAQUIM, and FDA was calculated using the Euclidean distance of the scaled properties. Besides, a CDP based on molecular fingerprints, scaffolds, and physicochemical attributes was utilized to assess the diversity of the datasets. The property-based diversity of the databases from PeruNPDB, BIOFAQUIM, and FDA was calculated using the Euclidean distance of the scaled properties. The values on the color CD plot are represented by data points on a continuous color scale. Brighter hues denote greater diversity, whereas darker colors denote less diversity. Last but not least, various point sizes are utilized to show the relative number of compounds in each database, with smaller data points representing databases with fewer molecules. Since it was discovered in the region where the greatest diversity in scaffold and fingerprints should be located, the findings showed that the compounds in the PeruNPDB had the greatest global diversity (Figure 3).
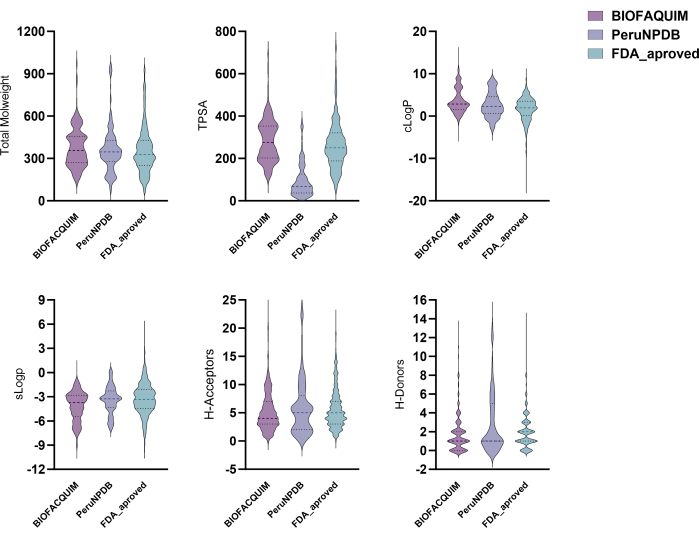
Figure 1: Violin plots for the physicochemical properties. Violin plots for the physicochemical properties of BIOFACQUIM, PeruNPDB, and FDA datasets. Please click here to view a larger version of this figure.
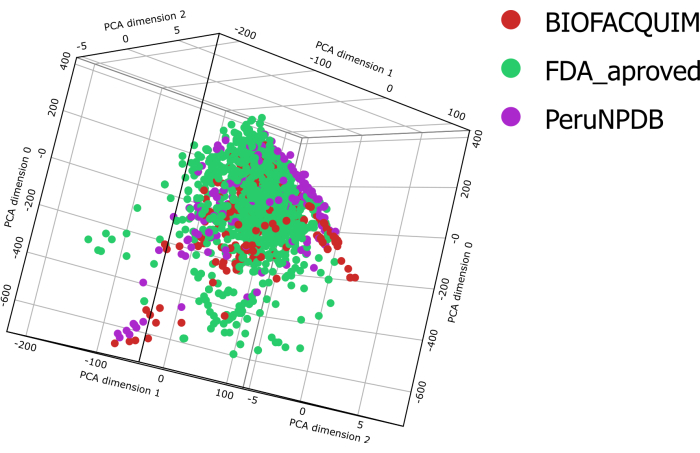
Figure 2: Visual representation of the chemical space. Visual representation of BIOFACQUIM, PeruNPDB, and FDA datasets based on principal components of six properties of pharmaceutical relevance. Please click here to view a larger version of this figure.
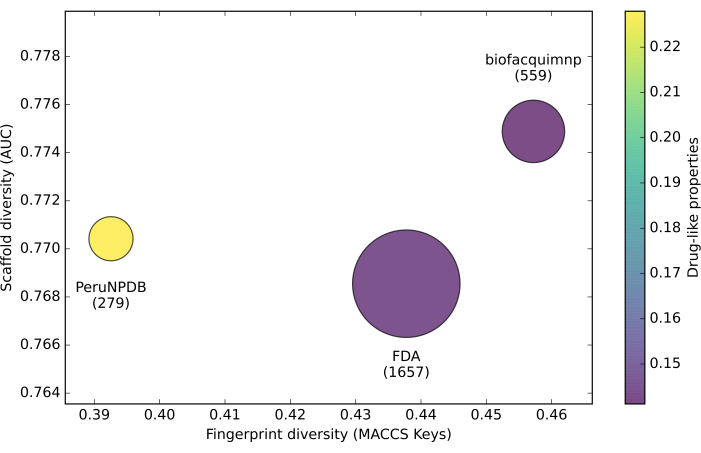
Figure 3: Consensus diversity plot. Consensus diversity plot comparing the global diversity of BIOFACQUIM, PeruNPDB, and FDA datasets. Please click here to view a larger version of this figure.
