By means of the described protocol, 3D PDMS cell culture substrates can be UV-photopatterned to create precise and high-throughput adhesive areas suitable for cell attachment. In this way, cells are subjected to both relevant substrate geometries and adhesive ligand patterns simultaneously. Cell properties such as orientation, cell area, and number of focal adhesions can easily be monitored and used to better understand cell behavior in complex, in vivo-like environments.
To verify the patterning events on the 3D PDMS substrates, atomic surface compositions of the material in different stages of the protocol have been measured using atomic X-ray photoelectron spectroscopy (XPS)48. In summary, the XPS measurements showed the presence of PEG chains with an increased carbon signal on passivated samples, which was reduced after photopatterning. Incubation with fibronectin resulted in an increase of carbon signal, again indicating successful protein adhesion on the surface of the cell culture chip. Next, pattern resolution and alignment on 3D features were characterized on a variety of circle-patterned, concave pits (ĸ = 1/250 µm-1, ĸ = 1/1,000 µm-1, and ĸ = 1/3,750 µm-1, see Figure 6). From the maximum-intensity projections, it can be concluded that the protein pattern was successfully patterned on all three 3D features. The intensity profile in Figure 6A shows high pattern resolution with sharp transitions between patterned and non-patterned areas. Additionally, consistent protein intensity across the complete pattern in the pit was obtained.
The concave pit with ĸ = 1/250 µm-1 was patterned using the single focal plane method (one pattern), whereas the pits with ĸ = 1/1,000 µm-1 and ĸ = 1/3,750 µm-1 were patterned using two and three focal planes (patterns), respectively. As can be seen in the maximum-intensity projections in Figure 6, both methods result in perfect alignment of the patterns on top of the features. No misaligned transitions between the two different focal planes and patterns can be observed.
Using the described protocol, a wide range of protein pattern designs can be applied to a variety of geometries (see Figure 7 and Video 1). To illustrate the versatility of this method, semicylinders (convex and concave), a saddle surface, and pit were patterned using lines and circles of various widths. The photopatterned materials can subsequently be used for cell culture (see Figure 7, Figure 8, Video 2, Video 3, and Video 4). An example of dermal fibroblasts cultured on a patterned (fibronectin lines, red, 5 µm wide, and 5 µm gaps) concave semicylinder is shown in Figure 8, Figure 9, and Video 4. During the experiment, cells sense and adhere to the multicue cell culture substrate and remain viable over time. As can be seen from the immunofluorescent staining in Figure 8, cells form focal adhesions (vinculin clusters) mainly on the fibronectin lines.
Another example study making use of these cell culture materials was recently published by our group48. In this study, human myofibroblasts and endothelial cells were subjected to the combination of contact guidance cues and geometrical topographies. In vivo, both types of cells experience curvature- and contact-guidance cues in native tissues such as in the human vasculature. By subjecting the cells in vitro to an environment that combines both environmental cues, the in vivo situation can be recapitulated, providing a deeper understanding of the role of the microenvironment on cell behavior. Human myofibroblasts were shown to align with contact guidance cues (parallel fibronectin lines) on concave cylindrical substrates48. However, on convex structures with increasing curvatures, the geometrical cues overruled the biochemical cues, suggesting that myofibroblasts can sense both the degree and sign of curvature. Interestingly, endothelial cells could only adhere to the concave multicue substrates and not to the convex PDMS substrates. On concave, protein-patterned substrates, the endothelial cells are oriented in the direction of the contact guidance cue. This fundamental in vitro knowledge has physiological relevance in the field of vascular tissue engineering and can eventually aid in the design of smart tissue engineering constructs.
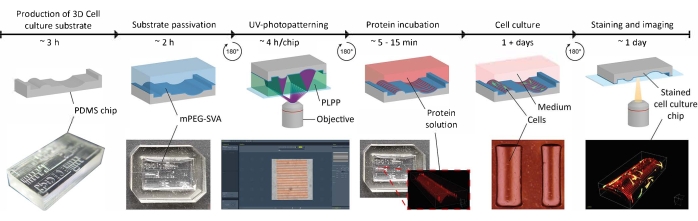
Figure 1: The experimental timeline of applying contact guidance cues on 3D cell culture substrates. First, positive cell culture chips are produced from a negative PDMS mold containing a range of geometries. Uncured PDMS is poured into the mold and cured for 3 h at 65 °C. Subsequently, the PDMS is treated with O2-plasma and incubated with PLL and mPEG-SVA (blue, labeled) to passivate the surface of the cell culture substrate. After washing, the substrate is flipped upside-down in a droplet of photoinitiator (PLPP, green, labeled) and UV-photopatterned using the LIMAP approach. Here, a digital mask with a user-defined pattern is used to cleave the passivation layer at defined locations. Next, a protein solution (red, labeled) can be incubated and will only adhere to the locations where the passivation layer is removed. Cells seeded on the substrate are subjected to both geometry and protein patterns, which enables research into cell behavior in complex, in vivo-mimicking environments. Abbreviations: PDMS = polydimethylsiloxane; mPEG-SVA = methoxypolyethylene glycol-succinimidyl valerate; PLPP = 4-benzoylbenzyl-trimethylammonium chloride; LIMAP = Light-Induced Molecular Adsorption of Proteins. Please click here to view a larger version of this figure.

Figure 2: Different stages during the production and passivation of the 3D cell culture substrate. The negative glass mold (#1) is designed with computer assisted design software and produced using a femtosecond-laser direct-write technique. This mold is utilized to produce the intermediate positive PDMS chip (#2) and negative PDMS mold (#3), which are subsequently used to produce the final cell culture chip (#4). Abbreviation: PDMS = polydimethylsiloxane. Please click here to view a larger version of this figure.
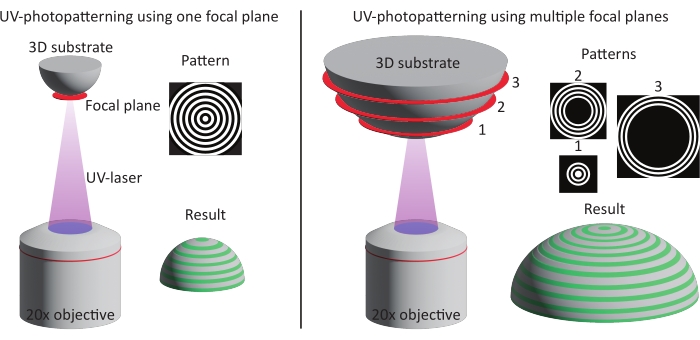
Figure 3: Schematic illustration of the two patterning methods. Left: UV-photopatterning is performed on smaller features (approximately one DMD) using a single focal plane and pattern. As a result, the complete feature is patterned in one go. Right: When larger features are used (larger than one DMD), the patterning is divided over multiple focal planes and patterns. Abbreviation: DMD = digital mirror device. Please click here to view a larger version of this figure.
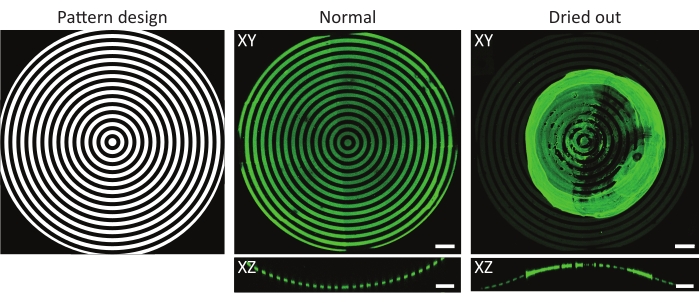
Figure 4: A typical example of a normal and dried-out protein-incubated substrate. Maximum-intensity projections (XY) and orthogonal views (XZ) of normal and dried-out protein-incubated substrates. When washing a patterned cell culture substrate after incubation with a protein solution, it is crucial to always keep the sample wet. Although the pattern is identical in all images on the features (ĸ = 1/1,000 µm-1), the gelatin-fluorescein (green) aggregated forming a major clump when the sample was left to dry for a few seconds. If the sample always remains wet, correct protein patterns can be observed. Scale bars = 100 µm. Please click here to view a larger version of this figure.

Figure 5: Brightfield images after seeding. Primary keratocytes (left) and dermal fibroblasts (right) 4 h after seeding on 3D geometrical features (concave pit of ĸ = 1/1,000 µm-1 and semicylinders of ĸ = 1/500, 1/375, 1/250, 1/175, and 1/125 µm-1). The top-left inserts represent the line pattern used for the patterning of the geometries. White arrows indicate spreading cells that already show alignment. Scale bars = 250 µm. Please click here to view a larger version of this figure.
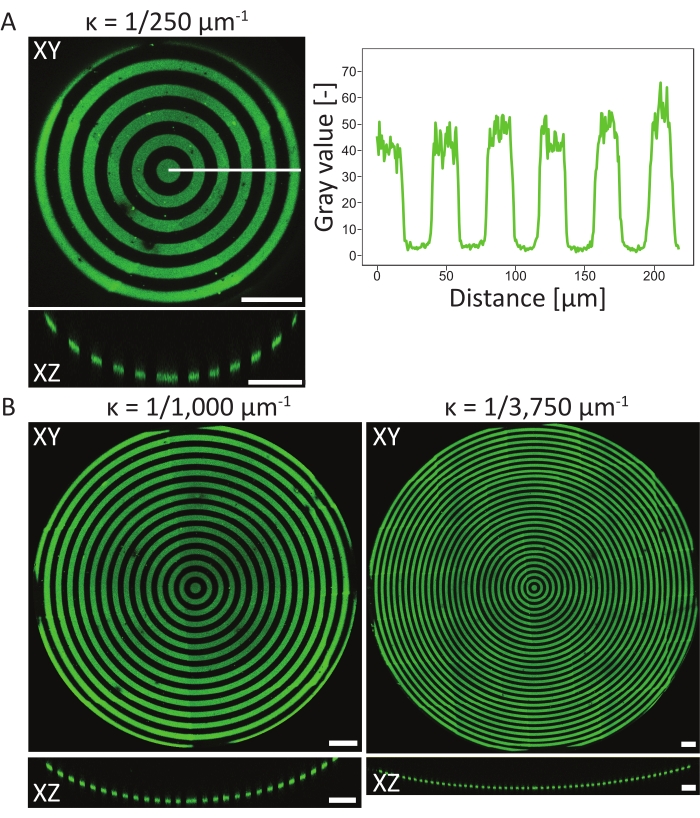
Figure 6: Characterization of circular patterns on concave pits. (A) The maximum-intensity projection (XY) and orthogonal view (XZ) of aconcave pit (ĸ = 1/250 µm-1) patterned using LIMAP (linewidth: 20 µm, gap width: 20 µm) and incubated with gelatin-fluorescein (green). The intensity profile along the white line is plotted against the distance, showing a consistent pattern quality and resolution. (B) Additional patterning performed on concave pits with ĸ = 1/1,000 µm-1 and ĸ = 1/3750 µm-1, showing flexibility in terms of geometrical features that can be used for patterning. Again, both the maximum intensity projections (XY) and orthogonal views (XZ) are visualized. Scale bars = 100 µm. Please click here to view a larger version of this figure.

Figure 7: 3D microscopy data of patterned structures. Typical examples of 3D patterned cell culture materials after photopatterning and cell culture, visualized using 3D rendering software. (A) Convex semicylinder patterned with 10 µm wide lines (rhodamine-fibronectin, red) and 10 µm wide gaps. Scale bar = 5 µm. (B) Dermal fibroblasts stained for F-actin (green) cultured on concave semicylinder patterned with 20 µm wide lines (rhodamine-fibronectin, red) and 20 µm wide gaps. Scale bar = 5 µm. (C) Saddle surface patterned with 20 µm wide lines (rhodamine-fibronectin, red) and 20 µm wide gaps. Scale bar = 5 µm. (D) Concave pit patterned with concentric circles of 20 µm wide lines (gelatin-fluorescein, green) and 20 µm wide gaps. The F-actin cytoskeleton of the human keratocytes is stained using phalloidin and visualized in red. Scale bar = 200 µm. Please click here to view a larger version of this figure.
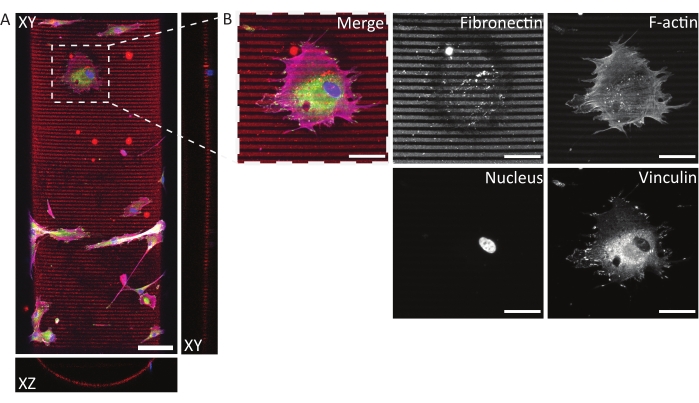
Figure 8: Immunofluorescent staining of human dermal fibroblasts on a photopatterned, concave semicylinder. (A) Maximum-intensity projection (XY) and orthogonal sections (XZ and YZ) of human dermal fibroblasts cultured for 24 h on a patterned (fibronectin lines, red, 5 µm wide, and 5 µm gaps) concave semicylinder. Cells are stained for F-actin (magenta), vinculin (green), and nuclei (blue). Scale bar = 100 µm. (B) Zoom-in of a cell adhering to the multicue environment. Scale bars = 50 µm. Please click here to view a larger version of this figure.

Figure 9: Brightfield timelapse images of human dermal fibroblasts on a patterned, concave cylinder. The concave semicylinder (ĸ = 1/250 µm-1) was patterned with parallel lines (5 µm wide and 5 µm gaps) and incubated with rhodamine-fibronectin before cell seeding. The timelapse imaging is started 1 h after initial cell seeding (left, 0 min), when cells are still rounded and non-adherent (arrows). After approximately 24 h (middle, 1,420 min), cells adhered to the multicue substrate and show an alignment response according to the contact guidance pattern. Both the alignment response and cell viability are maintained throughout the entire culture duration (right, 3,180 min). Scale bars = 200 µm. Please click here to view a larger version of this figure.
Video 1: Pattern example on a 3D cylindrical substrate. 3D representation of a convex cylinder patterned with rhodamine-fibronectin (red). Please click here to download this Video.
Video 2: 3D representation of dermal fibroblasts cultured on a patterned, 3D cylindrical substrate (ĸ = 1/500 µm-1). Dermal fibroblasts cultured for 24 h on a patterned (fibronectin lines, red, 10 µm wide, and 10 µm gaps) convex semicylinder. Cells are stained for F-actin (magenta), vinculin (green), and nuclei (blue). Please click here to download this Video.
Video 3: 3D representation of human keratocytes cultured on a patterned, 3D pit (ĸ = 1/3,750 µm-1). 3D representation of human keratocytes cultured for 24 h in a concave, patterned pit (gelatin circles, green, 20 µm wide, and 20 µm gaps). Cells are stained for F-actin (red). Please click here to download this Video.
Video 4: Brightfield timelapse imaging of human dermal fibroblasts on a patterned, concave cylinder. The concave semicylinder (ĸ = 1/250 µm-1) was patterned with parallel lines (5 µm wide and 5 µm gaps) and incubated with rhodamine-fibronectin before cell seeding. The timelapse imaging is started 1 h after initial cell seeding, when cells show initial adherence to the multicue environment. During the complete timelapse, cells predominantly orient along the contact guidance cues, while cell viability is maintained. Please click here to download this Video.
