1. Preparation of reagents for aniline tagging
- Prepare a 6 M aniline solution at pH 4.5. Working in a hood, combine 550 µL of aniline with 337.5 µL of LCMS grade water and 112.5 µL of 12 M hydrochloric acid (HCl) in a centrifuge tube. Vortex well and store at 4 °C.
NOTE: Aniline can be stored at 4 °C for 2 months.
CAUTION: Aniline is highly toxic and should be worked with in a fume hood. Hydrochloric acid is highly corrosive - Prepare a 6 M 13C aniline solution at pH 4.5. Combine 250 mg of 13C6-aniline with 132 µL of water and 44 µL of 12 M HCl. Vortex well and store at 4 °C.
- Prepare 200 mg/mL N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC) solution. Dissolve 2 mg of EDC in 10 µL of water for every sample to be tagged and vortex well.
NOTE: EDC solution should be prepared the same day as the reaction. EDC acts as a catalyst for the derivatization of compounds with aniline12.
2. Preparation of standards
- Make separate stock solutions of all compounds dissolved in LC/MS grade water (Table 1).
- Preparation of internal standard stock solution
- Combine all compounds except for nicotinamide adenine dinucleotide (NAD), nicotinamide adenine dinucleotide phosphate (NADP), flavin adenine dinucleotide (FAD), acetyl coenzyme A (ACA), and glycerol 3-phosphate (Gly3P), with the appropriate volumes to create a 2 mM stock solution of all compounds.
- Combine NAD, NADP, FAD, ACA, and Gly3P with the appropriate volumes to create a 2 mM stock solution.
3. Preparation of sample (Figure 1)
- Quench and precipitate the proteins in a cell-free protein synthesis reaction by adding an equal volume of ice-cold 100% ethanol to the reaction. Centrifuge the sample at 12,000 x g for 15 min at 4 °C. Transfer the supernatant to a new centrifuge tube.
NOTE: Samples can be stored at -80 °C at this point and analyzed at a later time
4. Labeling reaction
- Labeling sample with 12C-aniline solution
- Transfer 6 µL of sample into a new centrifuge tube and bring the volume to 50 µL with water.
NOTE: Volume sample size may depend on the specific CFPS reaction. - Add 5 µL of 200 mg/mL EDC solution.
- Add 5 µL of 12C-aniline solution.
NOTE: The aniline solution separates into two phases. Mix well before adding to the reaction. - Vortex the reaction with gentle shaking for 2 h at room temperature.
- After 2 h, remove the tubes from the shaker and add 1.5 µL of triethylamine (TEA) to the reaction in a fume hood.
NOTE: Triethylamine raises the pH of the solution which stops the aniline tagging reaction and stabilizes the compounds.
CAUTION: Triethylamine is toxic and causes irritation of the eyes and respiratory tract. - Centrifuge at 13,500 x g for 3 min.
- Transfer 6 µL of sample into a new centrifuge tube and bring the volume to 50 µL with water.
- Labeling internal standards with 13C-aniline solution
- Dilute internal stock solution to 80 µM with a final volume of 50 µL.
NOTE: Concentration of internal standards can be adjusted to levels close to the experimental sample. - Add 5 µL of 200 mg/mL EDC solution.
- Add 5 µL of 13C-aniline solution.
- Vortex the reaction with gentle shaking for 2 h at room temperature.
- After 2 h, remove the tubes from the shaker and add 1.5 µL of TEA to the reaction in a fume hood.
- Centrifuge at 13,500 x g for 3 min.
- Dilute internal stock solution to 80 µM with a final volume of 50 µL.
- Combining tagged internal standard and tagged sample
- Mix 25 µL of 12C-aniline labeled sample with 25 µL of 13C-aniline labeled standard.
- Transfer to an auto-sampler vial and analyze by the LC/MS procedure.
- Creating a standard curve for untagged metabolites
- Dilute stock solution of untagged metabolites (NAD, NADP, FAD, ACA, and Gly3P) to final concentrations of 320 µM, 80 µM, 20 µM and 5 µM with a volume of 50 µL.
- Add 5 µL of 200 mg/mL EDC solution.
- Add 5 µL of 12C-aniline solution.
- Vortex the reaction with gentle shaking for 2 h at room temperature.
- After 2 h, remove the tubes from the shaker and add 1.5 µL of TEA to the reaction in a fume hood.
- Centrifuge at 13,500 x g for 3 min.
- Transfer supernatant to an auto-sampler vial and analyze by the LC/MS procedure.
NOTE: The untagged metabolites follow the same procedure as the sample to replicate the sample matrix in order to maintain similar ionization efficiency.
5. Setup of LC/MS procedure
- Preparation of solvents
- Prepare 5 mM tri-butylamine (TBA) aqueous solution adjusted to pH 4.75 with acetic acid.
NOTE: TBA in the mobile phase helps the analytes achieve good resolution and separation14. - Prepare 5 mM TBA in acetonitrile (ACN).
- Prepare wash solvent with 5% water and 95% ACN.
- Prepare purge solvent with 95% water and 5% ACN.
- Prepare 5 mM tri-butylamine (TBA) aqueous solution adjusted to pH 4.75 with acetic acid.
- Setup of MS conditions
- Set the mass spectrometer to negative ion mode with a probe temperature of 520 °C, negative capillary voltage of -0.8 kV, positive capillary voltage of 0.8 kV, and set the software to acquire data at 5 points/s.
- Set selected ion recordings (SIR) for each metabolite with specified cone voltages and mass over charge (m/z) values. See Table 1.
- Initializing LC/MS according to manufacturer’s instructions
- Prime solvent lines in the solvent manager for 3 min.
- Prime wash solvent (5% water, 95% ACN) and purge solvent (95% water, 5% ACN) for 15 s for 5 cycles.
- Set the sample manager to 10 °C.
- Install a C18 (1.7µm, 2.1mm x 150mm) column and initialize column with 100% ACN at 0.3 mL/min for 10 min.
- Condition the column at 95% water and 5% ACN at 0.3 mL/min for 10 min prior to introducing solvents with buffers.
- Condition the column at 95% solvent A (5mM TBA aqueous, pH 4.75) and 5% solvent B (5 mM TBA in ACN) at 0.3 mL/min for 10 min.
- Set up a gradient protocol with the elution starting at 95% solvent A and 5% solvent B, raised to 70% solvent B in 10 min, raised to 100% solvent B in 2 min and held at 100% solvent B for 3 min. Return to initial conditions (95% solvent A, 5% solvent B) over 1 min and hold for 9 min to re-equilibrate the column.
- Condition the column with the gradient protocol 3 times prior to any injections onto the column.
- Injecting sample and standards
- Inject 5 µL of the sample into the column and acquire the appropriate m/z ion intensities for the 12C-aniline tagged sample.
- Inject 5 µL of the same sample again, but this time acquire the m/z ion intensities for the 13C-aniline tagged standards.
NOTE: Our LC/MS system is unable to acquire both 12C and 13C m/z intensities at the specified SIR time windows, since it is too much data to acquire in the specified time window. Therefore, we inject the same sample twice. - Inject untagged metabolite standards from lowest concentration to highest and record the appropriate m/z ion intensities.
6. Quantification
- Creating Export method
- In data acquisition software, select File > New Method > Export Method.
- Specify a Filename, such as AnilineTagging_Date.
- Check the Export ASCII File and choose a directory to export the text file to.
- In Report Type, select Summary by All.
- In Delimiters, for Column select a ,. For Row, select [cr][if].
- In Table, select Export and then Edit Table to include SampleName, Area, Alto, Amount and Units.
- Save export method.
- Quantifying metabolites with internal standards using data acquisition software
- Under the Sample Sets tab, right click the corresponding LC/MS run and select View as > Channels.
- Select all SIR channels for the 13C-aniline internal standards of one injection, right click and select Review.
- If the LC Processing Method Layout window does not automatically appear, go to View > Processing Method Layout.
- In Processing Method Layout, go to the Integration tab and set ApexTrack as the algorithm.
- Go to the Smoothing tab and set the type to Mean and the smoothing level to 13.
NOTE: Any smoothing level can be selected, as long as it is consistent across all samples. - In the MS Channel tab, disable MS 3D Processing.
- In the SIR channel window, integrate each peak, one channel at a time. Once a peak is integrated, go to Options > Fill from Result and the details of the peak will be filled in the Components tab. Change the peak name to the corresponding compound name.
- Once all the SIR channels have been evaluated, save the processing method and close window.
- Select all SIR channels of the 13C-aniline and 12C-aniline tagged sample, right click and select Process.
- Check the Process box, select Use specified processing method, and choose the processing method that is just saved. Also check the Export box, select Use specified export method and choose the saved export method created earlier. Click OK.
- Open the exported text file with Excel and calculate the concentration of the unknown compound using:
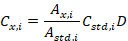
where Cx,i is the concentration of the unknown sample for metabolite i, Ax,i is the integrated area of the unknown metabolite i, Astd,i is the integrated area of the internal standard of metabolite i, Cstd,i is the concentration of the internal standard of metabolite i, and D is the dilution factor.
- Quantifying untagged metabolites with standard curve
- Under the Sample Sets tab, right click the corresponding LC/MS run and select View as > Channels.
- Select all SIR channels for the untagged standards of one injection, right click and select Review.
- If the LC Processing Method Layout window does not automatically appear, go to View > Processing Method Layout.
- In Processing Method Layout, go to the Integration tab and set ApexTrack as the algorithm.
- Go to the Smoothing tab and set the type to Mean and the smoothing level to 13.
NOTE: Any smoothing level can be selected, as long as it is consistent across all samples. - In the MS Channel tab, disable MS 3D Processing.
- In the SIR channel window, integrate each peak, one channel at a time. Once a peak is integrated, go to Options > Fill from Result and the details of the peak will be filled in the Components tab. Change the peak name to the corresponding compound name.
- Once all the SIR channels have been evaluated, save the processing method and close window.
- Under the Sample Sets tab, right click on the sample set and select Alter Sample.
- Select Amount in the new window.
- Select copy from Process method and choose the process method that was just saved.
- Enter the concentration of each metabolite for each vial and enter the unit as <μM for each component (or the corresponding unit) and select OK.
- Select the sample set again, right click, View as > Channels.
- Select all SIR channels of the untagged metabolites for the standards, right click and select Process.
- Check the Process box and choose Use specified processing method. Select the appropriate processing method and click OK.
- Select SIR channels for all untagged metabolites for the samples, right click and select Process.
- Check the Process box, select Use specified processing method, and choose the processing method that was just saved. Also check the Export box, select Use specified export method and choose the saved export method created earlier. Click OK.
- Quantify the untagged metabolites with the standard curve and export the results to a text file to the directory specified.
As a proof-of-concept, we used the protocol to quantify metabolites in an E. coli based CFPS system expressing green fluorescent protein (GFP). The CFPS reaction (14 μL) was quenched and deproteinized with ethanol. The CFPS sample was then tagged with 12C-aniline, while standards were tagged with 13C-aniline. The tagged sample and standards were then combined and injected into the LC/MS (Figure 1). The protocol detected and quantified 40 metabolites involved in central carbon and energy metabolism using internal standards, while a standard curve for 5 of the metabolites that were not tagged with aniline was also developed (Figure 2). The diverse metabolites involved in these pathways were a class of phosphorylated sugars, phosphocarboxylic acids, carboxylic acids, nucleotides, and cofactors. The derivatization with aniline introduced a hydrophobic moiety into hydrophilic molecules which facilitated more effective separation using reversed-phase chromatography12. In addition, the method enabled the separation of structural isomer pairs such as glucose 6-phosphate and fructose 6-phosphate in a single LC/MS run. Each compound’s mass over charge (m/z) ratio and retention time were identified prior to the experiment by injecting 1 mM of one compound at a time and comparing the mass spectrum to the blank (Table 1).
The limit of detection and range of linearity for all compounds was estimated by producing a standard curve that ranged from 0.10 µM to 400 µM (Table 2). The average correlation coefficient (R2) for all compounds was 0.988 and most compounds had a linear range of 3-orders of magnitude. Three compounds had notable saturation effects, especially alpha-ketoglutarate which had a linear range from 0.1 µM to 25 µM. Isocitrate and citrate also had saturation effects above 100 µM.
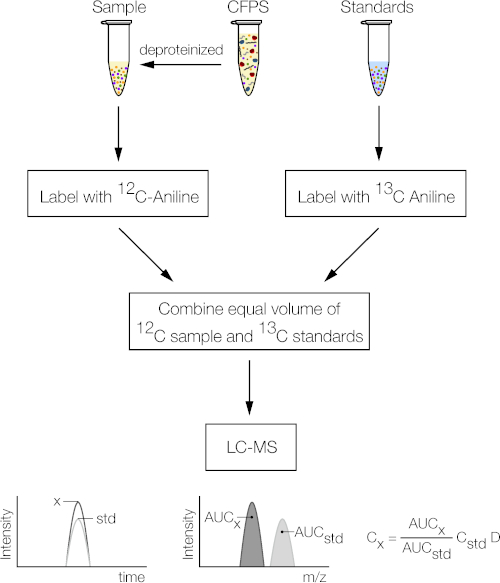
Figure 1: Schematic of workflow for aniline tagging. The cell-free protein synthesis reaction is deproteinized and tagged with 12C-aniline, while a standard stock mixture is tagged with 13C-aniline. Both mixtures are then mixed at a 1:1 volumetric ratio and analyzed by LC/MS. Please click here to view a larger version of this figure.
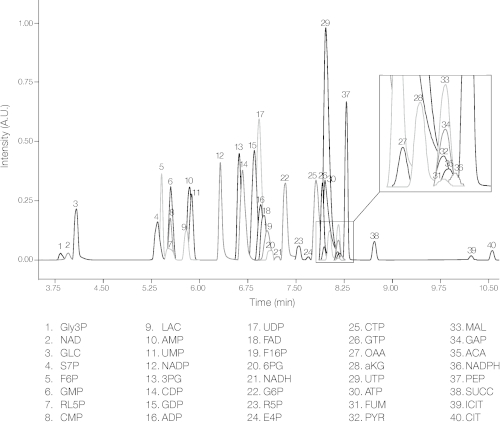
Figure 2: Overlapped selected ion chromatograms for 40 metabolites. Mass chromatogram from a single LC/MS run of a 40 µM standard mixture of 40 metabolites. Peaks were identified by their retention time and m/z values for each compound. Complete compound names and their abbreviations are listed in Table 1. Please click here to view a larger version of this figure.
| Peak Number | Metabolite | Abbreviation | KEGG ID | Retention Time (min) | 12C m/z | 13C m/z | nonlabel m/z | CV | MS Species | |
| 1 | Glycerol 3-phosphate | Gly3P | C00093 | 3.85 | 153 | 10 | M – H2O – H | |||
| 2 | Nicotinamide adenine dinucleotide | NAD | C00003 | 3.96 | 698 | 10 | M + Cl – H | |||
| 3 | Glucose | GLC | C00031 | 4.06 | 289.9 | 296 | 15 | M + A + Cl – H | ||
| 4 | Sedoheptulose 7-phosphate | S7P | C05382 | 5.41 | 364 | 370 | 10 | M + A – H | ||
| 5 | Fructose 6-phosphate | F6P | C00085 | 5.48 | 334 | 340 | 10 | M + A – H | ||
| 6 | Guanosine monophosphate | GMP | C00144 | 5.57 | 437.05 | 443 | 10 | M + A – H | ||
| 7 | Ribulose 5-phosphate | RL5P | C00199 | 5.58 | 304 | 310 | 10 | M + A – H | ||
| 8 | Cytidine monophosphate | CMP | C00055 | 5.59 | 397.09 | 403 | 10 | M + A – H | ||
| 9 | Lactate | LAC | C00186 | 5.77 | 164.05 | 170 | 10 | M + A – H | ||
| 10 | Adenosine monophosphate | AMP | C00020 | 5.85 | 421.1 | 427.1 | 10 | M + A – H | ||
| 11 | Uridine monophosphate | UMP | C00105 | 5.88 | 398.07 | 404 | 10 | M + A – H | ||
| 12 | Nicotinamide adenine dinucleotide phosphate | NADP | C00006 | 6.39 | 724 | 10 | M – H2O – H | |||
| 13 | 3-Phosphoglyceric acid | 3PG | C00197 | 6.63 | 242 | 248.06 | 15 | M + A – H2O – H | ||
| 14 | Cytidine diphosphate | CDP | C00112 | 6.72 | 477 | 483 | 10 | M + A – H | ||
| 15 | Guanosine diphosphate | GDP | C00035 | 6.87 | 517 | 523 | 10 | M + A – H | ||
| 16 | Adenosine diphosphate | ADP | C00008 | 6.94 | 501 | 507 | 10 | M + A – H | ||
| 17 | Uridine diphosphate | UDP | C00015 | 6.97 | 478 | 484 | 10 | M + A – H | ||
| 18 | Flavin adenine dinucleotide | FAD | C00016 | 7.03 | 784.15 | 15 | M – H | |||
| 19 | Fructose 1,6-bisphosphate | F16P | C05378 | 7.1 | 395.95 | 402.1 | 10 | M + A – H2O – H | ||
| 20 | Gluconate 6-phosphate | 6PG | C00345 | 7.11 | 425.1 | 437 | 10 | M + 2A – H | ||
| 21 | Nicotinamide adenine dinucleotide reduced | NADH | C00004 | 7.23 | 633.13 | 639.08 | 10 | M + A + H2O – nicotinamide – H | ||
| 22 | Glucose 6-phosphate | G6P | C00668 | 7.32 | 409.1 | 421.1 | 10 | M + 2A – H | ||
| 23 | Ribose 5-phosphate | R5P | C00117 | 7.54 | 379.1 | 391.1 | 15 | M + 2A – H | ||
| 24 | Erythrose 4-phosphate | E4P | C00279 | 7.71 | 348.9 | 361 | 10 | M + 2A – H | ||
| 25 | Cytidine triphosphate | CTP | C00075 | 7.84 | 557 | 563 | 5 | M + A – H | ||
| 26 | Guanosine triphosphate | GTP | C00044 | 7.93 | 597 | 603 | 5 | M + A – H | ||
| 27 | Oxalacetate | OAA | C00036 | 7.94 | 281 | 293 | 25 | M + 2A – H | ||
| 28 | Alpha-ketoglutarate | aKG | C00026 | 7.95 | 295 | 307.1 | 15 | M + 2A – H | ||
| 29 | Uridine triphosphate | UTP | C00075 | 7.97 | 558 | 564 | 10 | M + A – H | ||
| 30 | Adenosine triphosphate | ATP | C00002 | 8.03 | 581 | 587 | 15 | M + A – H | ||
| 31 | Fumarate | FUM | C00122 | 8.09 | 265 | 277.1 | 10 | M + 2A – H | ||
| 32 | Pyruvate | PYR | C00022 | 8.09 | 162 | 168 | 25 | M + A – H | ||
| 33 | Malate | MAL | C00149 | 8.09 | 283.06 | 295.15 | 10 | M + 2A – H | ||
| 34 | D-glyceraldehyde 3-phosphate | GAP | C00118 | 8.09 | 319 | 331.1 | 5 | M + 2A – H | ||
| 35 | Acetyl-coenzyme A | ACA | C00024 | 8.16 | 790 | 10 | M – H2O – H | |||
| 36 | Nicotinamide adenine dinucleotide phosphate reduced | NADPH | C00005 | 8.23 | 694.92 | 700.82 | 10 | M + A – nicotinamide – H | ||
| 37 | Phosphoenolpyruvate | PEP | C00074 | 8.28 | 317 | 329.1 | 20 | M + 2A – H | ||
| 38 | Succinate | SUCC | C00042 | 8.64 | 267.07 | 279.1 | 15 | M + 2A – H | ||
| 39 | Isocitrate | ICIT | C00311 | 10.13 | 398 | 416 | 10 | M + 3A – H2O – H | ||
| 40 | Citrate | CIT | C00158 | 10.46 | 416.1 | 434.06 | 20 | M + 3A – H | ||
Table 1: Identification and labeling results of metabolites. Each compound’s corresponding peak number, retention time, m/z value for unlabeled, 12C and 13C labeled, and MS species. MS Species, A stands for Aniline tag.
| Peak No. | Metabolite | Abbreviation | KEGG ID | Concentration (mM) | SD (n = 3) | Limit of Detection (μM) | Limit of Linear Range (μM) | R^2 | |
| 1 | Glycerol 3-phosphate | Gly3P | C00093 | 0.377 | 0.034 | 0.1 | 400 | 0.995 | |
| 2 | Nicotinamide adenine dinucleotide | NAD | C00003 | 0.052 | 0.010 | 0.39 | 400 | 0.993 | |
| 3 | Glucose | GLC | C00031 | 0.002 | 0.000 | 0.1 | 400 | 0.997 | |
| 4 | Sedoheptulose 7-phosphate | S7P | C05382 | 0.007 | 0.000 | 0.16 | 400 | 0.988 | |
| 5 | Fructose 6-phosphate | F6P | C00085 | 0.029 | 0.004 | 0.1 | 400 | 0.986 | |
| 6 | Guanosine monophosphate | GMP | C00144 | 0.007 | 0.001 | 0.39 | 100 | 0.992 | |
| 7 | Ribulose 5-phosphate | RL5P | C00199 | 0.035 | 0.002 | 0.39 | 400 | 0.996 | |
| 8 | Cytidine monophosphate | CMP | C00055 | 0.045 | 0.001 | 0.1 | 100 | 0.992 | |
| 9 | Lactate | LAC | C00186 | 2.134 | 0.048 | 0.1 | 400 | 0.988 | |
| 10 | Adenosine monophosphate | AMP | C00020 | 0.020 | 0.002 | 0.1 | 100 | 0.992 | |
| 11 | Uridine monophosphate | UMP | C00105 | 0.021 | 0.000 | 0.1 | 100 | 0.997 | |
| 12 | Nicotinamide adenine dinucleotide phosphate | NADP | C00006 | 0.014 | 0.002 | 0.34 | 400 | 0.950 | |
| 13 | 3-Phosphoglyceric acid | 3PG | C00197 | 6.125 | 0.239 | 0.1 | 100 | 0.996 | |
| 14 | Cytidine diphosphate | CDP | C00112 | 0.202 | 0.029 | 0.39 | 400 | 0.997 | |
| 15 | Guanosine diphosphate | GDP | C00035 | 0.146 | 0.027 | 1.5625 | 400 | 0.984 | |
| 16 | Adenosine diphosphate | ADP | C00008 | 0.797 | 0.161 | 0.39 | 400 | 0.995 | |
| 17 | Uridine diphosphate | UDP | C00015 | 0.212 | 0.036 | 0.39 | 400 | 0.991 | |
| 18 | Flavin adenine dinucleotide | FAD | C00016 | 0.008 | 0.001 | 0.1 | 400 | 0.958 | |
| 19 | Fructose 1,6-bisphosphate | F16P | C05378 | 3.643 | 0.105 | 0.39 | 400 | 0.989 | |
| 20 | Gluconate 6-phosphate | 6PG | C00345 | 0.017 | 0.001 | 0.39 | 400 | 0.989 | |
| 21 | Nicotinamide adenine dinucleotide reduced | NADH | C00004 | 0.063 | 0.028 | 0.39 | 100 | 0.972 | |
| 22 | Glucose 6-phosphate | G6P | C00668 | 0.046 | 0.002 | 0.1 | 400 | 0.984 | |
| 23 | Ribose 5-phosphate | R5P | C00117 | 0.055 | 0.005 | 0.39 | 100 | 0.999 | |
| 24 | Erythrose 4-phosphate | E4P | C00279 | 0.038 | 0.007 | 0.39 | 400 | 0.979 | |
| 25 | Cytidine triphosphate | CTP | C00075 | 0.896 | 0.078 | 6.25 | 100 | 0.998 | |
| 26 | Guanosine triphosphate | GTP | C00044 | 0.870 | 0.109 | 6.25 | 100 | 0.993 | |
| 27 | Oxalacetate | OAA | C00036 | 0.023 | 0.008 | 0.56 | 400 | 0.997 | |
| 28 | Alpha-ketoglutarate | aKG | C00026 | 0.391 | 0.020 | 0.1 | 25 | 0.979 | |
| 29 | Uridine triphosphate | UTP | C00075 | 0.845 | 0.092 | 1.5625 | 400 | 0.998 | |
| 30 | Adenosine triphosphate | ATP | C00002 | 1.557 | 0.188 | 1.5625 | 400 | 0.991 | |
| 31 | Fumarate | FUM | C00122 | 0.576 | 0.100 | 1.5625 | 100 | 0.999 | |
| 32 | Pyruvate | PYR | C00022 | 5.813 | 0.804 | 0.39 | 400 | 0.993 | |
| 33 | Malate | MAL | C00149 | 2.548 | 0.269 | 0.1 | 400 | 0.991 | |
| 34 | D-glyceraldehyde 3-phosphate | GAP | C00118 | 2.194 | 0.367 | 0.1 | 100 | 0.974 | |
| 35 | Acetyl-coenzyme A | ACA | C00024 | 0.196 | 0.044 | 0.1 | 100 | 0.991 | |
| 36 | Nicotinamide adenine dinucleotide phosphate reduced | NADPH | C00005 | 0.006 | 0.010 | 0.14 | 100 | 0.990 | |
| 37 | Phosphoenolpyruvate | PEP | C00074 | 3.442 | 0.345 | 0.1 | 100 | 0.962 | |
| 38 | Succinate | SUCC | C00042 | 5.683 | 0.573 | 0.1 | 320 | 0.999 | |
| 39 | Isocitrate | ICIT | C00311 | 0.003 | 0.006 | 0.39 | 100 | 0.998 | |
| 40 | Citrate | CIT | C00158 | 0.002 | 0.001 | 0.1 | 100 | 0.981 | |
Table 2: Metabolite quantification in a representative CFPS sample. The concentration of each metabolite and the standard deviation. Limit of detection, range of linearity and correlation coefficient identified from standard curves.
