We first validated the viability of embryos imaged using the protocol's parameters for diSPIM acquisition (sections 1-6). Ten embryos were simultaneously imaged at 20 °C, one volume/embryo/minute, from the 2-cell stage to the 2-fold stage (7.5 hours, 451 volumes/embryo). To monitor cell divisions throughout embryogenesis, we used strain BV514, which ubiquitously expresses the mCherry::Histone reporter constructs from the integrated transgene array ujIs11324. Figure 6 shows a timeline of this first half of embryonic development for one of the imaged embryos. Each image represents a single-view maximum-intensity projection (produced by steps 7-8) of the imaged embryo. We found that the optimized protocols did not induce any detectable phototoxicity to the embryos, as assessed by timing of cell divisions (not shown), time of hatching, and timing related to developmental milestones (Figure 6 and references1,25,26).
We then applied the protocol to analyze outgrowth dynamics of single neurons in developing embryos. We imaged DCR7692 (olaex4655), a transgenic nematode strain that expresses GFP off the neuropeptide flp-19 promoter in a subset of unidentified cells (DACR2819, Pflp-19 (3.6kb)::Syn21::GFP::CAAX::p10 3’UTR). Following the steps of the protocol outlined here, we determined that the unidentified cells correspond to motor neurons RMDDL and RMDDR, to the excretory canal cell, and to two muscle cells (Figure 7). We then examined and quantified the outgrowth dynamics of the RMDDL and RMDDR neurons. We observed that the RMDDL and RMDDR neurons are obliquely shaped as early as 360 minutes post fertilization, with the longer cellular axis representing the subsequent axis for neurite outgrowth (Figure 7 and Movie S1). Using the “simple neurite tracing” plugin in FIJI and applying it to 3D reconstructions of isotropically fused volumes, we then quantified the stereotypic outgrowth of the RMDDL and RMDDR neurites for six embryos. We determined that the outgrowth dynamics were stereotyped for RMDDL and RMDDR across embryos (herein called RMDDs). From 385-410 minutes post fertilization, the RMDDs neurites extended 6.0±0.5µm (mean ± SEM; n = 12 neurites) anterior of the cell bodies (Figure 7B,C,I). From 415-445 minutes post fertilization, both neurites extend dorsally into and around the presumptive nerve ring (asterisk in Figure 7D). On average, each RMDD neurite extended 11.0 ± 0.6 µm (mean ± SEM; n = 12 neurites) from the cell body before synchronously meeting its contralateral counterpart at the apex of the ring (Figure 7I). Importantly, our representative results demonstrate that we are able to examine, compare and quantify neuronal developmental features for single identifiable cells by using our integrated protocol (Figure 7 and Figure 8).
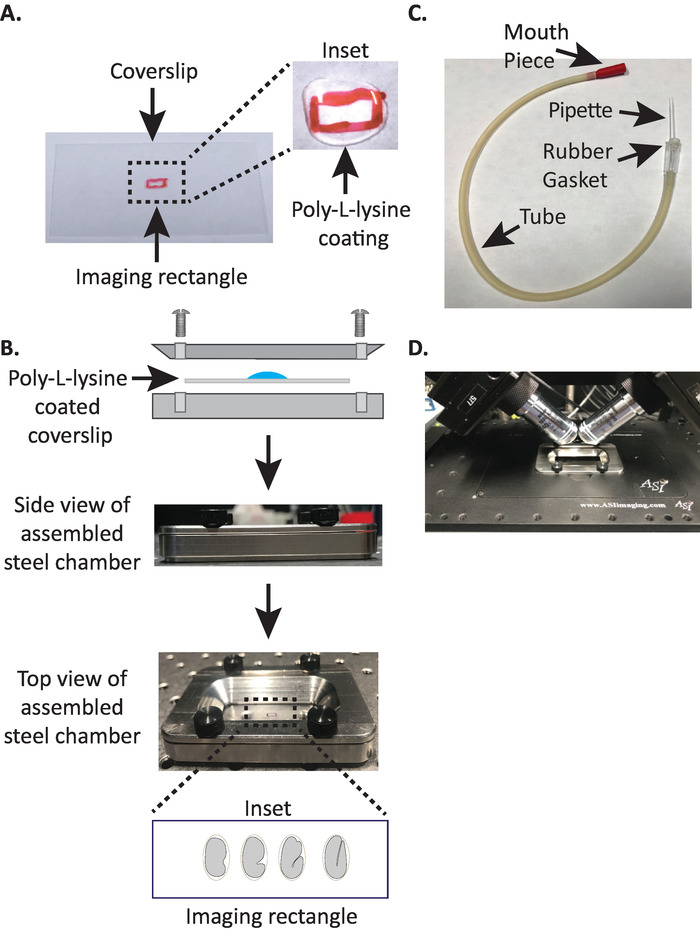
Figure 1: diSPIM sample mounting setup. (A) Preparation of coverslip with Poly-L-lysine. In the inset, 10 µL of Poly-L lysine was used to coat the coverslip for 5 minutes. Poly-L-lysine allows the embryo eggshell to stick firmly to the coverslip in the rectangle. (B) Schematic of the steel imaging chamber and the assembled chamber. In the inset, representative embryos are oriented with the anterior-posterior axis perpendicular to the long axis on the coverslip. (C) Assembled aspirator tube with microcapillary pipette. (D) Steel imaging chamber mounted in sample holder under diSPIM 40x objectives. Please click here to view a larger version of this figure.
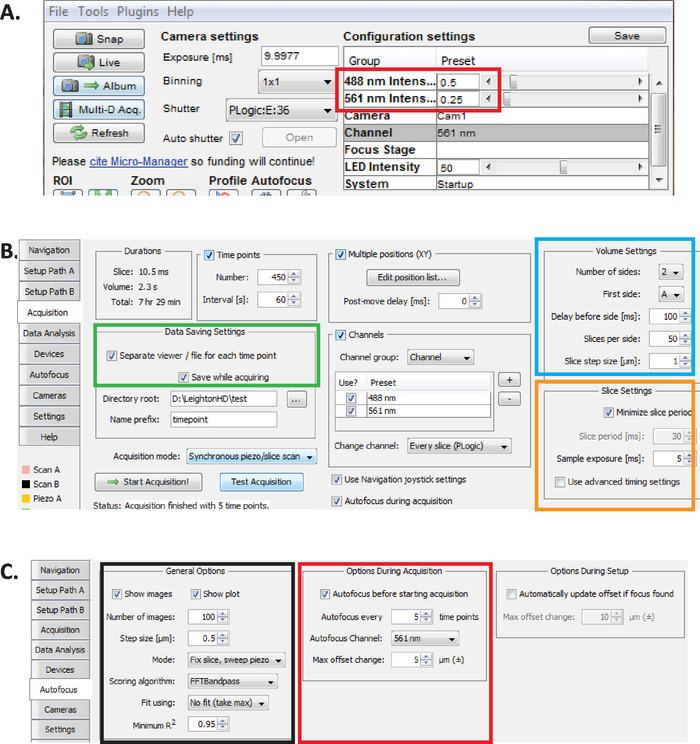
Figure 2: Long-term diSPIM imaging setup in Micro-Manager. (A) Recommended diSPIM laser power parameters (red rectangle) optimized for prolonged imaging while reducing phototoxicity (as assessed by higher hatching rate of C. elegans embryos). Set 561 nm laser to 79 µW (0.25) and 488 nm laser to 179 µW (0.5). Note that exact calibration of software settings to laser power varies among diSPIM installations. It is recommended that users measure and calibrate the laser power in order to achieve 79 µW (561 nm) and 179 µW (488 nm) laser power. (B) diSPIM parameters for data saving (green rectangle), volume settings (blue square), and slice settings (orange square). (C) diSPIM autofocus parameters for long-term imaging of C. elegans embryogenesis (see steps 6.1-6.6). Please click here to view a larger version of this figure.

Figure 3: Image visualization and data-processing setup using CytoSHOW. (A) Raw diSPIM images opened by CytoSHOW. CytoSHOW is able to open images captured by both camera paths (SPIM A and B). These raw images are opened in multidimensional windows called diSPIM monitor. In diSPIM monitor, a “bowtie pattern” is generated to select the embryo’s anterior, posterior, dorsal and ventral edges (see step 9.1). Bow-tie selections indicate embryo orientation for deconvolution and StarryNite-assisted lineaging tracing. (B) Optimized parameters used to generate isotropic images. In the Deconvolved while acquiring window, set the parameters specified in steps 9.5.1-9.5.8. Please click here to view a larger version of this figure.
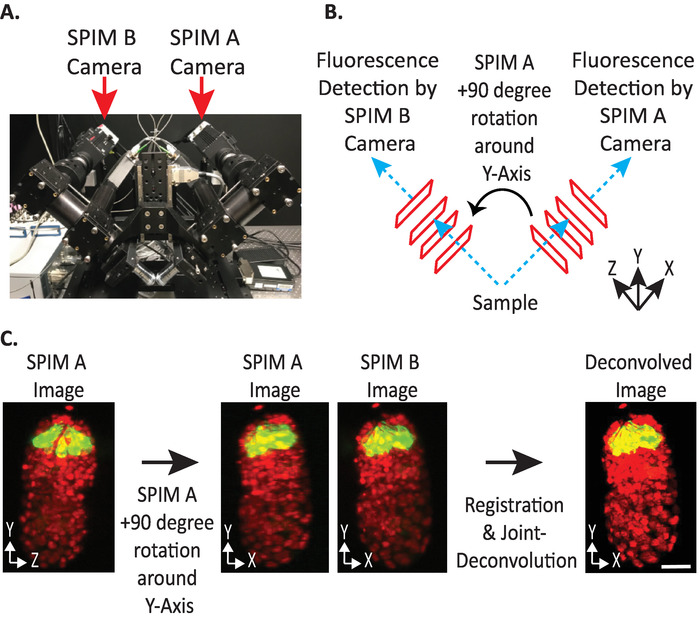
Figure 4: diSPIM camera configuration. (A) Photograph of diSPIM camera placements and orientations. (B) Depiction of +90-degree rotations of SPIM A to match SPIM B images collected. (C) Input volumes relative to orientation index +1 based on our diSPIM’s camera configuration (see step 9.5.2). We rotate SPIM A image(s) +90 degrees around Y-Axis before registration to match SPIM B image(s). Scale bars = 10 µm. Images are representative single-view, maximum-intensity projections and deconvolution images of 1.5-fold embryo with labeled nuclei (561-nm, red) and neurons (488-nm, green). Please click here to view a larger version of this figure.
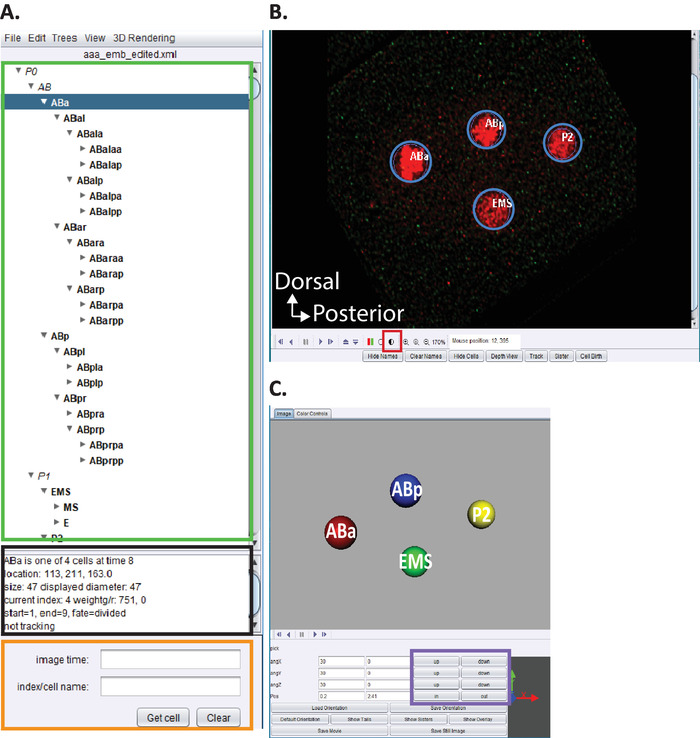
Figure 5: Curation and editing C. elegans embryonic lineage in AceTree. (A) We use AceTree to edit StarryNite’s lineage traces (see references5,6,8; manuscripts are also included in our download bundle). AceTree will display C. elegans systematic names for each nucleus (green rectangle) upon completion of steps 10.1-10.2. This window (A) provides information (black rectangle) about each cell in the lineage trace (ABa, highlighted in blue) that help guide users when tracking and editing the lineage traces. It is recommended that users verify and compare lineaged cells and their positions to the C. elegans embryonic cell lineage previously reported by Sulston et al.1 In addition, if users are interested in locating specific cells in the deconvolved data series (see below, B) enter the C. elegans systematic name in the search bar (orange rectangle). (B) The user’s deconvolved data series also opens automatically upon completion of steps 10.1-10.2. Shown here an isotropically fused image of a four-cell stage embryo with nuclei labeled in red. During tracking of a nuclei, users should change the intensity of the image (red square) and navigate through time and z using the arrow keys on their keyboard (time-left/right, z-up/down). (C) 3D cartoon of the timepoint in (B) with certain functionalities (purple rectangle) that enables rotatable 3D-visualization. For an overview of AceTree and its 3D functionality, see references5,6,8. Please click here to view a larger version of this figure.
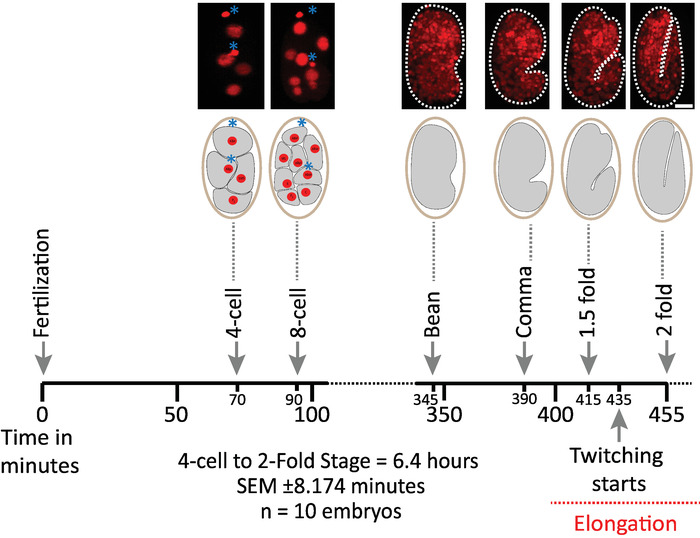
Figure 6: Timed developmental dynamics of C. elegans embryos on the diSPIM. Top panel, diSPIM images showing the first half of embryonic development for one of the imaged embryos (Strain BV514 ujIs11324). Embryos were imaged continuously, every minute for 7.5 hours (at 20 °C). The first two images of top panel represent 4- and 8-cell embryos with nuclei (red) and positions of polar bodies (dense red spheres, marked with blue asterisks). Each image represents a single-view maximum-intensity projection of the imaged embryo. Scale bars = 10 µm. The timeline (horizontal bar) represents minutes post fertilization (m.p.f.) of the development of C. elegans embryos. We validated that our protocol's parameters for diSPIM acquisition did not induce any detectable phototoxicity to the imaged embryos as assessed by viability, timing of cell divisions, timing of hatching and timing of developmental milestones (see references1,25,26). We note that the timing of developmental milestones was reproducible across embryos with our imaging parameters (SEM ± 8.174 minutes for 6.4 hour long imaging sessions; n = 10). Please click here to view a larger version of this figure.
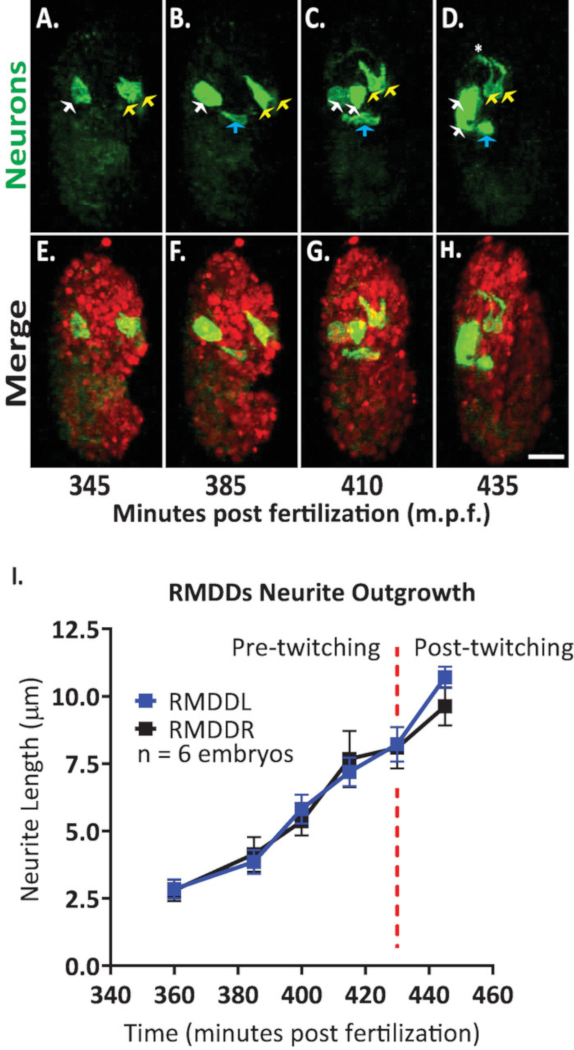
Figure 7. Cellular identification and single cell characterization of neurite outgrowth dynamics in developing C. elegans embryos. Dual-color imaging of a strain made by crossing BV514 ujIs11324 (for lineaging) and DCR7692 (olaex4655), a transgenic nematode strain that expresses GFP off the neuropeptide flp-19 promoter in a subset of unidentified cells. (A-H) Following the steps of the protocol outlined here, we determined that the unidentified cells correspond to motor neurons RMDDL and RMDDR (yellow arrows), to the excretory canal cell (blue arrows), and to two muscle cells (white arrows). (I) Quantification of the outgrowth dynamics of the RMDDL and RMDDR neurons by using FIJI plugin “simple neurite tracing” and applying it to 3D reconstructions of isotropically fused volumes. Note how both RMDDL and RMDDR show stereotypic outgrowth dynamics, each extending synchronously for a total length of 11.0±0.6µm (mean ± SEM; n = 12 neurites) and meeting at the dorsal apex of nerve ring (see also Movie S1). Please click here to view a larger version of this figure.
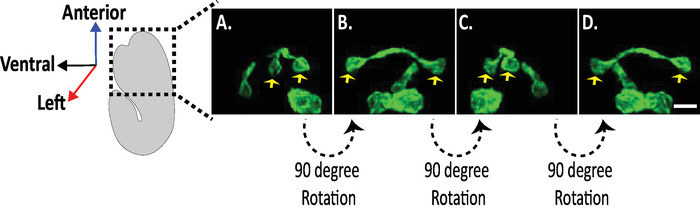
Figure 8: Examination of Isotropic diSPIM images of neuronal morphologies in C. elegans embryos. Isotropic visualization of AVHL and AVHR neurons (yellow arrows). Using the diSPIM, neuronal morphologies can be captured yielding four-dimensional (4D) images with isotropic spatial resolution of approximately 330 nm. The diSPIM allows users to virtually rotate image volumes with identical resolution in all direction. Images in A–D are maximum-intensity projections of the same isotropically fused diSPIM image volume from distinct rotations around the embryo's long axis. Scale bars = 5 µm. Please click here to view a larger version of this figure.
Supplementary Movie S1: C. elegans embryo developing from 280 to 434 minutes post fertilization. Isotropic movie of strain DCR7692 (olaex4655) expressing ujIs113 ubiquitously with DACR2819 sparsely labeling RMDD neurites (Figure 7A-D, yellow arrows). DACR2819 also labels two muscle cells (Figure 7A-D, white arrows) and excretory canal cell (Figure 7A-D, blue arrow) during embryonic development (Figure 7A-D). Scale bars = 10 µm. Please click here to download this file.
