Circadian bioluminescence reporter vector: human PER2 promoter-driven expression of destabilized luciferase variant
The DNA sequence comprising a 941 bp fragment derived from the human PER2 promoter used to construct the circadian reporter vector, pGL[hPer2P/Luc2P/Neo, was first analyzed for the presence of regulatory elements known to regulate circadian gene expression. Bioinformatics analysis showed that within this promoter fragment, there are three different Bmal1 binding sites (E-box) (CAT/CGTG), two circadian transcription enhancing sites (CCAATG), two GC boxes, and a transcription start site (Figure 1). Hence this reporter vector was anticipated to reflect circadian gene expression.
Optimization of synchronization agent
Several agents have been used to synchronize or affect the autonomous circadian rhythm of fibroblast cells for in vitro bioluminescence studies. To select an efficient and cost-effective cell-type specific synchronization agent in mammary epithelial cells, we compared 10 µM forskolin, 1 nM melatonin, 0.1 µM dexamethasone, 50% HS, and 100% SQ in mammary epithelial cells (MCF10A) transiently transfected with the circadian reporter vector. Cells synchronized with 50% HS showed the best circadian rhythm, with 3 peaks of circadian induced luminescence, compared to only one or two peaks in cells synchronized with other agents. In addition, the bioluminescence profiles of cells synchronized with 50% HS produced acceptable period, amplitude, and best-fit value during data analysis (Figure 2). Based on these findings, we selected this cost-effective method as the cell synchronization agent for all subsequent experiments.
Disruption and restoration of cellular circadian rhythm by chemical exposures
In this in vitro model, untreated, transiently transfected cells (control group) showed at least two complete cycles of luminescence signaling after synchronization (Figure 3A). Cells exposed to the direct acting mutagen, NMU, showed a dose-dependent disruption of circadian gene expression as reflected by the loss of luminescence peaks. While treatment with 0.25 mM NMU did not disrupt the cellular circadian rhythm, a dose of 0.5 mM NMU initially delayed and later abolished circadian rhythm, as indicated by the disappearance of the second peak of luminescence at ~72 h post-treatment. Inhibition of SIRT1 activity with 20 nM Ex257 or 1 µM cambinol similarly disrupted circadian rhythm by dampening the subsequent circadian cycle (Figure 3A-1). Importantly, the addition of MSC (12.5 µM) to the culture medium restored towards normal first and second cycles of circadian gene expression in NMU-treated cells. MSC not only restored the rhythm disrupted by NMU, but also prevented the disruptive effects of SIRT1 inhibitors, Ex257 and cambinol (Figure 3A-2).
In stably transfected cells, both NMU and SIRT1 inhibitors disrupted circadian rhythm, albeit at higher concentrations than that observed in transient transfectants (Figure 3B-1). MSC (12.5 µM) restored circadian rhythms in the cells pretreated with 1 mM NMU (Figure 3B-2). More importantly, MSC also restored circadian rhythms in the cells treated with SIRT1 inhibitors, including EX257 (40 nM) (Figure 3B-3) and cambinol (2 µM) (Figure 3B(4)), respectively.
In comparison between Figure 3A and Figure 3B, the bioluminescence intensity was much higher but the rhythm was sustained for a shorter time in transiently transfected cells vs stably transfected cells (~ 10 times higher, 2 times shorter) in general, even after treatment of cells with NMU or MSC. In addition, the cellular circadian rhythm was 2-fold more sensitive to exposure to chemicals in transiently transfected cells compared to the stably transfected cells.
Quantitative dose-dependent change of cellular circadian rhythm by chemicals
Similarly, the disrupted cellular circadian rhythm of the stably transfected cells treated with 1 mM NMU was restored by the treatment of NAC in a dose-dependent manner (0-20 µM) (Figure 4A) as observed in the changes of circadian period (Figure 4B) and phase (Figure 4C).
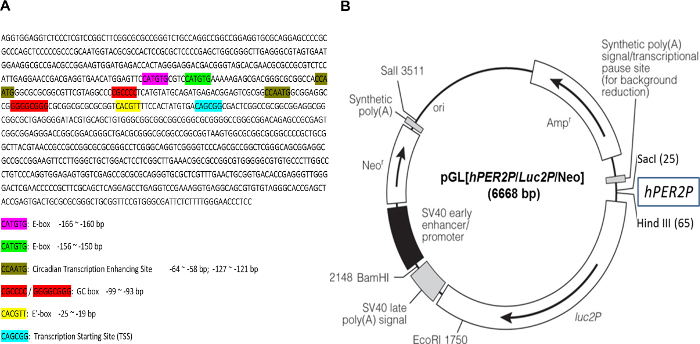
Figure 1: Sequence of human PER2 promoter fragment and the circadian reporter vector. (A) Human PER2 promoter fragment (941 bp) was sequenced, and the sequence was analyzed for the elements of functional circadian promoter. E-box, CACGTT / CATGTG; circadian transcription enhancing site, CCAATG; GC box, CGCCCC / GGGGCGGG; transcription starting site (TSS): CAGCGG. (B) Schematic diagram of destabilized luciferase reporter vector, pGL[hPer2P/Luc2P/Neo]. The transcription of destabilized luciferase (dLuc) is under direct control of the hPER2 promoter. A co-expressed neomycin resistance gene (Neo) facilitates selection of infected cells by G418. Please click here to view a larger version of this figure.
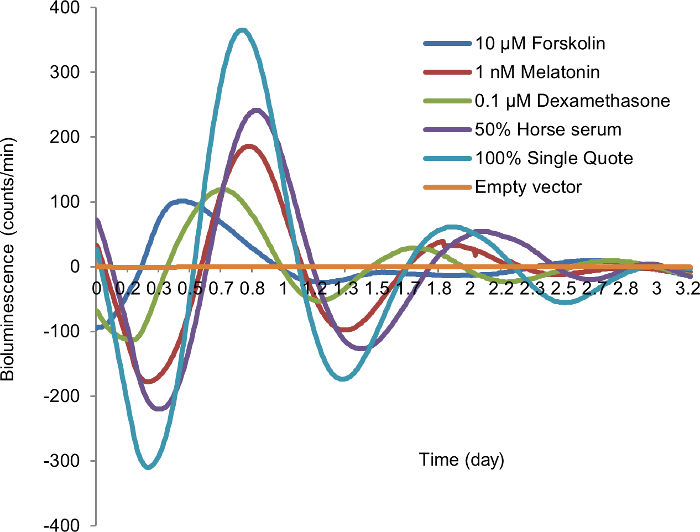
Figure 2: Synchronization of in vitro cellular circadian rhythm by different synchronization agents. After 24 h starvation, MCF10A cells transiently transfected by circadian reporter vector were treated with each synchronization agent for 2 h, followed by recording luminescence signal. Blue, 10 µM forskolin; red, 1 nM melatonin; green, 0.1 µM dexamethasone; purple; 50% horse serum (HS); teal, 100% SingleQuot (SQ); yellow, empty vector (negative control). X-axis, time (day); Y-axis, bioluminescence (count/min). Please click here to view a larger version of this figure.
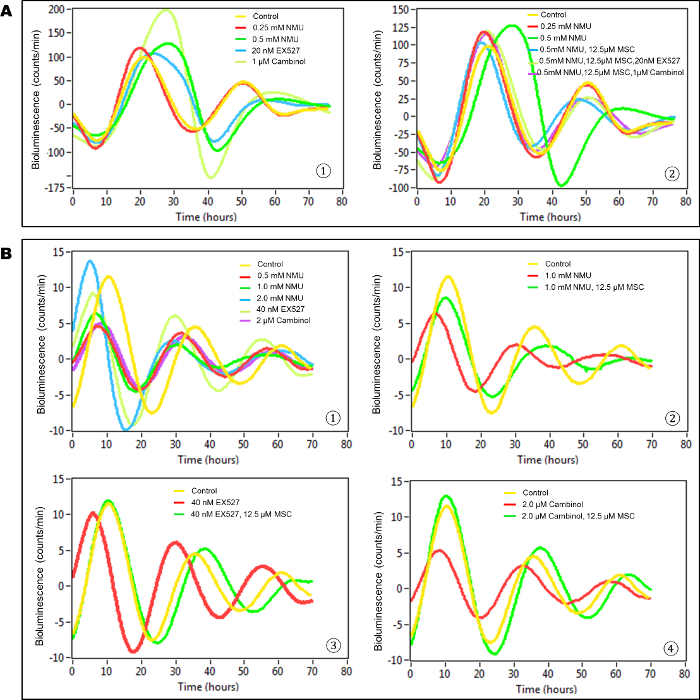
Figure 3: MSC restored cellular circadian rhythms disrupted by NMU and SIRT1 inhibitors in mammary epithelial cells in vitro. Bioluminescence assays were performed on MCF10A/PER2-dLuc reporter cells after synchronization with 50% HS. X-axis, time (h) (post-NMU treatment time); Y-axis, bioluminescence (count/min). (A) Results from transiently transfected cells. (1) Cells were treated with 0, 0.25 or 0.5 mM NMU, 20 nM EX527, or 1 µM cambinol for 1 h following synchronization. Yellow: control; red: 0.25 mM NMU; green: 0.5 mM NMU; blue: 20 nM EX527; yellow green: 1µM cambinol. (2) Cells were treated with 12.5 µM MSC alone, or in combination with 20 nM EX527 or 1 µM cambinol in recording medium following exposure to 0.5 mM NMU. Yellow: Control; red: 0.25 mM NMU; green: 0.5 mM NMU; blue: 0.5 mM NMU + 12.5 µM MSC; yellow green: 0.5 mM NMU + 12.5 µM MSC + 20 nM EX527; purple: 0.5 mM NMU + 12.5 µM MSC + 1 µM cambinol. (B) Results from stably transfected cells. The 3rd– 5th peaks which showed clean and clear differences among groups are presented as a representative result. (1) Cells were treated with 0, 0.5, 1.0 or 2.0 mM NMU, 40 nM EX527, or 2 µM cambinol for 1 h after synchronization. Yellow: Control; red: 0.5 mM NMU; green: 1.0 mM NMU; blue: 2.0 mM NMU; yellow green: 40 nM EX527; purple: 2 µM cambinol. (2) Cells were treated with 12.5 µM MSC in recording medium following exposure to 1.0 mM NMU. Yellow: Control; red: 1.0 mM NMU; green: 1.0 mM NMU + 12.5 µM MSC. (3) Cells were treated with 12.5 µM MSC following exposure to 40 nM EX257. Yellow: Control; red: 40 nM EX257; green: 40 nM EX257 + 12.5 µM MSC. (4) Cells were treated with 12.5 µM MSC following exposure to 2 µM cambinol. Yellow: Control; red: 2 µM cambinol; green: 2 µM cambinol + 25 µM MSC. This result was previously reported and is licensed under CC BY 2.022. Please click here to view a larger version of this figure.
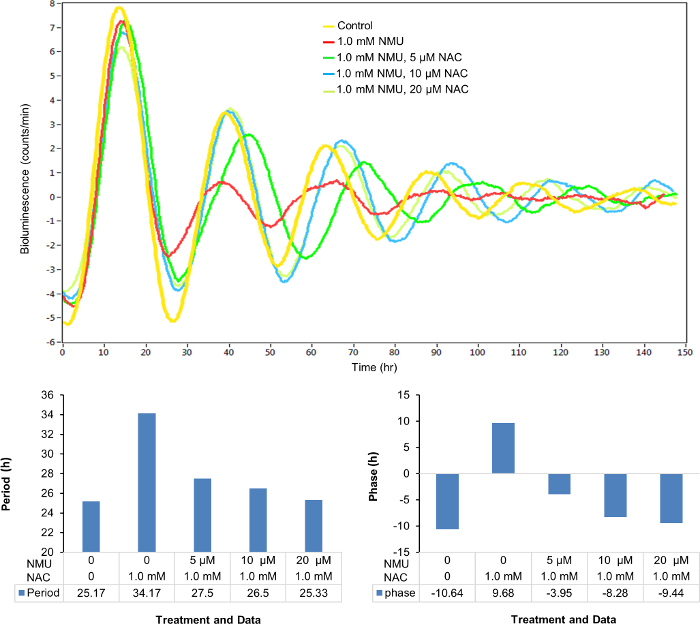
Figure 4: NAC dose-dependently restored the cellular circadian rhythm disrupted by NMU. Cells were treated with 1.0 mM NMU or vehicle control for 1 h after synchronization. (A) Representative result of bioluminescence (count/min) against time (h). Yellow: Control; red: 1.0 mM NMU; green: 1.0 mM NMU, 5 µM NAC; blue: 1.0 mM NMU, 10 µM NAC; yellow green: 1.0 mM NMU, 20 µM NAC. (B) Period (hours). (C) Phase (hours). Please click here to view a larger version of this figure.
