In Ovo Electroporation in the Chicken Auditory Brainstem
Summary
Auditory brainstem neurons of avians and mammals are specialized for fast neural encoding, a fundamental process for normal hearing functions. These neurons arise from distinct precursors of embryonic hindbrain. We present techniques utilizing electroporation to express genes in the hindbrain of chicken embryos to study gene function during auditory development.
Abstract
Electroporation is a method that introduces genes of interest into biologically relevant organisms like the chicken embryo. It is long established that the chicken embryo is an effective research model for studying basic biological functions of auditory system development. More recently, the chicken embryo has become particularly valuable in studying gene expression, regulation and function associated with hearing. In ovo electroporation can be used to target auditory brainstem regions responsible for highly specialized auditory functions. These regions include the chicken nucleus magnocellularis (NM) and nucleus laminaris (NL). NM and NL neurons arise from distinct precursors of rhombomeres 5 and 6 (R5/R6). Here, we present in ovo electroporation of plasmid-encoded genes to study gene-related properties in these regions. We show a method for spatial and temporal control of gene expression that promote either gain or loss of functional phenotypes. By targeting auditory neural progenitor regions associated with R5/R6, we show plasmid transfection in NM and NL. Temporal regulation of gene expression can be achieved by adopting a tet-on vector system. This is a drug inducible procedure that expresses the genes of interest in the presence of doxycycline (Dox). The in ovo electroporation technique – together with either biochemical, pharmacological, and or in vivo functional assays – provides an innovative approach to study auditory neuron development and associated pathophysiological phenomena.
Introduction
Fast neural encoding of sound is essential for normal auditory functions. These include sound localization abilities1, speech in noise discrimination2, and the comprehension of other behaviorally relevant communication signals3. Analogous neurons located in the auditory brainstem of both avians and mammals are highly specialized for fast neural encoding4. These include the chicken nucleus magnocellularis (NM), the nucleus laminaris (NL) and their mammalian analogs, the anteroventral cochlear nucleus (AVCN) and the medial superior olive (MSO), respectively5. However, developmental mechanisms regulating fast neural encoding are poorly understood in the auditory brainstem. Therefore, it is advantageous to study specific genes that are responsible for fast neural encoding in order to better understand their expression, regulation and function in auditory development.
The developing chicken embryo is an effective and well-established research tool to study basic biological questions of auditory system development6,7. Recent molecular advances have addressed these biological questions in the developing chicken embryo by expressing or knocking down genes of interest in order to analyze in vivo gene function8,9. Investigating the regulatory role of specific genes is a significant advancement in understanding pathologies associated with auditory deficits. Here, we present in ovo electroporation of plasmid-encoded genes into the chicken auditory brainstem where fast neural encoding of sound occurs10. By targeting auditory neural progenitor regions associated with rhombomeres 5 and 611,12 (R5/R6), we show spatial control of plasmid transfection in NM and NL. In addition, we show temporal regulation of expression by adopting a tet-on vector system. This is a drug inducible procedure that expresses the genes of interest in the presence of doxycycline (Dox)8.
Protocol
All procedures were approved by Northwestern University Institutional Animal Care and Use Committees, and carried out in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
1. Egg Handling
- Purchase fertilized eggs from a local vendor. Store eggs at 13 °C in a refrigerator for no longer than 5 days prior to incubation. Embryo viability significantly decreases after 1 week.
- Swab each egg with 70% ethanol before setting in the incubator.
- Set 1-2 dozen(s) of eggs on their sides in an incubator with the temperature set to 38 °C at ~50% humidity. This ensures proper embryo positioning for electroporation and optimal viability, respectively.
NOTE: After 48-52 h of incubation, the eggs will be at Hamburger-Hamilton (HH) stage ~12-13 and ready for electroporation.
2. Preparation
- Plasmid Mixing
- Add 1 µL of 0.1% fast green (0.1% in 18 MΩ deionized and distilled H2O [ddH2O]) for every 7 µL of DNA mixture.
- For co-electroporation of multiple plasmids, mix plasmids in a 1:1 ratio. The final DNA concentration should be 5-8 µg/µL.
- Fill Injection Pipette
- Pull pipettes on a micropipette puller. Fill the pipette with 1-2 µL plasmids using a 28G syringe needle.
- Break pipette tip to 10 – 20 µm using forceps. Use a microscope to assist in determining the size of the broken pipette tip. Attach the pipette to the pipette holder of the picospritzer.
3. Windowing
NOTE: The windowing procedure has been published before13. Please see reference 13 for additional visual information.
- Wipe all instruments and the work area with 70% ethanol.
- Take the desired number of set eggs out of the incubator (between 6-10), leave at room temperature, and individually swab eggs with 70% ethanol again. Eggs remain viable for at least 2 h after incubator removal.
- Place the egg on top of a light illuminator to identify embryo positioning. Circle the center of the dark area (i.e., embryo) with a pencil.
- Using a 19G needle, poke a hole in the blunt end of the egg.
- Poke another hole near the drawn circle on the pointed side of the egg. Remove a small piece of the outer eggshell (about 16 mm2) with the 19G needle and then remove a small piece of the inner eggshell (about 8 mm2) with forceps.
- With a 3 mL syringe fitted with a 19G needle, withdraw 1.5-2.5 mL of albumin from the egg through the blunt-end hole. Be sure to angle the needle down at 45°.
- Cover the blunt-end hole with a small piece of tape (1 cm x 1 cm). Cover the drawn circle and the second hole with tape (2.5 cm x 4 cm).
- With curved scissors (cutting edge = 2.5 cm), cut a window within the circle. Scissors should be parallel to the eggshell and the diameter of the window should be less than 2 cm.
4. Plasmid Injection
- Turn on the picospritzer. Set the pressure to 18 psi and the duration to 5 µs. Turn on the air tank and lamp for the dissection microscope.
- Make 30 mL stock solution of a 1:10 dilution of store-bought Indian ink mixed in sterile phosphate-buffered saline (PBS). Store at 4 °C. Injection of Indian ink is an important step to get a better visualization of the chicken embryo.
- Fill a 1 mL syringe with the diluted Indian ink solution. Attach a 27G needle and expel any bubbles that are present in the syringe.
- Insert needle just under the yolk membrane ~2 – 3 mm from the embryo and inject ~0.2 mL of Indian ink gently under the embryo. Do not inject more than 0.5 mL of the Indian ink as it may decrease embryo survival.
- Place the windowed egg on an egg holder under the dissection microscope. Use the highest magnification for best visualization of plasmid injection site.
- Using forceps, remove the membrane over the injection area. Auditory brainstem regions of interest (i.e., NM and NL) arise from Rhombomeres 5 and 6 (R5/R6). R5/R6 are located adjacent to the otocysts (an embryonic structure in vertebrates that develops into the inner ear) that serve as landmarks for R5/R6 plasmid injections.
- Lower the DNA-filled pipette into the neural tube overlying the R5/R6 region and between otocysts using the micromanipulator. A schematic of ideal placement is shown in Figure 1A.
- Apply air pressure (18 psi, 5 µs duration) from the picospritzer to eject DNA in the neural tube.
5. Electroporation
- Turn on the current/voltage stimulator.
- Fill a 3 mL syringe with PBS and attach the syringe filter. Add 1-2 drops of PBS onto the embryo.
- Lower the bipolar electrode to the embryo using the micromanipulator with the negative electrode above the injection site (medial) and the positive electrode lateral to the R5/R6 (Figure 1A). Avoid direct contact with the embryo. Use bipolar electrodes where the electrode material is platinum iridium. Adjust the spacing between the tips to 0.5 mm with forceps.
- Using a current/voltage stimulator, pulse 20 times at 50 V for 1 ms at 1 s intervals.
- After electroporation, add one drop of PBS over the exposed area. Gently remove the electrode and clean it with a laboratory tissue and 70% ethanol. Close the window exposing the embryo with tape.
- Label eggshell with the injected plasmid type and the hatch date.
- Place eggs back into incubator with window side up. Incubate at 38 °C with 50% humidity until desired developmental stage is reached.
- If a tet-on vector is electroporated for temporal control of gene expression, apply Doxycycline (Dox) every 24 h to induce gene expression (see Section 6).
6. Tet-on System for Temporal Control of Gene Expression
- Use the following plasmids:
pCAGGS-T2TP: a transposase expressing plasmid.
pT2K-CAGGS-rtTA-M2: a plasmid that stably expresses the Dox-binding protein.
pT2K-BI-TRE-EGFP: a plasmid that contains a bidirectional tet-on promoter (TRE)-driving expression of the EGFP reporter and a second gene of interest.
NOTE: The above three plasmids need to be co-electroporated in a 1:1:1 ratio. - Stock solution preparation:
- Dissolve Dox in sterile PBS to produce a 1 mg/mL stock solution. Store at -20 °C.
- Dox application:
- To induce the expression of the gene of interest during certain developmental stages, apply Dox every 24 h.
- When applying Dox, take the egg out, open the window, pipette 50 µL Dox onto the chorioallointoic membrane, close the window and put the egg back into the incubator.
7. Preparation of Dissecting Area for Brainstem Slices
NOTE: The following sections (7 – 9) and procedures have been published before14. Please see these references for additional visual information.
- Prepare an agar solution (40 mg/mL agarose, i.e. 4% in ddH2O). Pour agar solution into a Petri dish and allow it to solidify at room temperature. Store covered agar plate in the refrigerator for future use during brainstem slicing of tissue.
- Continually bubble artificial cerebral spinal fluid (ACSF) with 95% O2 and 5% CO2 (pH 7.2-7.4 and osmolarity: 295-310 mOsm/L).
- Clean the working area and vibratome with 70% EtOH and rinse the blade with distilled water.
- Place a clean fluid absorption pad on the working area with appropriate dissecting tools.
- Take the agar plate out of the refrigerator, cut a small agar cube (10 mm x 10 mm x 8 mm). Glue the agar block to the stage of a vibratome-slicing chamber using commercially available superglue.
8. Isolation of Chicken Auditory Brainstem
- Open the egg at taped window site. Puncture the membrane sac with a scalpel and remove the head of the chicken embryo.
- Decapitate the chicken with sharp scissors.
- Place the razor blade tip slightly posterior to the eyes. Incise through the skull at the rostral-to-caudal midline. Apply slight pressure depending on the age of the embryo. Older embryos require more pressure.
- Gently push aside skin and feathers to expose skull and verify midline cut.
- Applying strong pressure, slice the rostral portion of skull with a razor blade. Immediately place blade posterior to eyes and cut through entire skull and brain tissue.
- Make midline-to-lateral incisions with scissors in the skull"s caudal region, slightly anterior to neck muscles on both sides of the head. Expose brain and cerebellum by pulling away skull and excess tissue.
- Cut tissue attached to the brainstem with scissors and remove the brainstem, which freely detaches from the skull.
9. Preparation of Brainstem Slices for In Vivo Electrophysiology or Imaging
- Pin down brainstem through the optic tecta.
- Remove the cerebellum by cutting peduncles with scissors, exposing the floor of the fourth ventricle.
- Using tweezers, remove any membranous tissue and blood vessels from the brainstem surface.
- To block the brainstem, make a horizontal cut at the most rostral end of the floor of the fourth ventricle, just caudal to the cortex. Make lateral cuts perpendicular through the optic tecta to isolate brainstem.
- Place a small amount of super glue on the vibratome stage directly in front of the agar block.
- Lift the brainstem at the spinal cord using tweezers and place it on the super glue with rostral side down and dorsal side towards the vibratome blade. Remove excess glue with a laboratory tissue or filter paper.
- Pour oxygenated ACSF into the vibratome stage. Set the vibratome blade at a 20-22° angle. Start vibratome at maximum-amplitude oscillation. Move the stage up towards the blade so the top of the tissue is parallel with the blade.
- Rapidly move blade towards brainstem tissue. Slow down the blade considerably prior to the blade"s contact with tissue.
- Slice tissue with the slowest possible forward speed. Once the blade is through the entire coronal tissue section, gently remove the slice using a transfer pipette.
- Lower the stage 200-300 µm and slice again. Repeat until anatomical landmarks of auditory nuclei become visible, e.g., the distinct dorsal/ventral neuropil region of NL and the medial location of NM relative to NL.
- Carefully maintain slices in ACFS. This can be done at room temperature (22° C) or near physiological conditions (~41° C) using a warm water bath. Note the order of slicing. The first slice corresponds to the most caudal region of tissue and the last slice corresponds to the most rostral region. This roughly represents the tonotopic organization of the auditory brainstem, from low- to high-frequency encoding regions, respectively.
Representative Results
We show here that in ovo electroporation permits gene expression in a normally developing biological system. Plasmid-encoded genes are focally injected into the neural tube overlying R5/R6. A schematic example of the electrode and pipette placements relative to important anatomical markers is shown in Figure 1A. The correct location of plasmid injection is confirmed 24 h after electroporation and shown in Figure 1B. The targeted injection of plasmids into the neural tube overlying R5/R6 permits expression in specific auditory brainstem regions, namely NM and NL(11). Figure 1C shows an in vitro brainstem slice from an E12 embryo under differential interference contrast (DIC) illumination. The auditory regions of interest are outlined. With florescent illumination (Figure 1C1), YFP expressing neurons within NM and NL are visible. Figure 1D shows DIC illumination for an E18 embryo under high magnification. The classic adendritic cell bodies of numerous NM neurons are apparent. With florescent illumination (Figure 1D1), an YFP expressing NM neuron is clearly visible while other transfected neurons are out of the focal plane. Because only a fraction of neurons in the targeted NM region are transfected, it allows other neurons within the same nucleus to serve as internal controls during dual patch-clamp electrophysiology experiments. Transgenes can also be temporally regulated by drug application, enabling gene expression and manipulation at precise developmental time periods8.
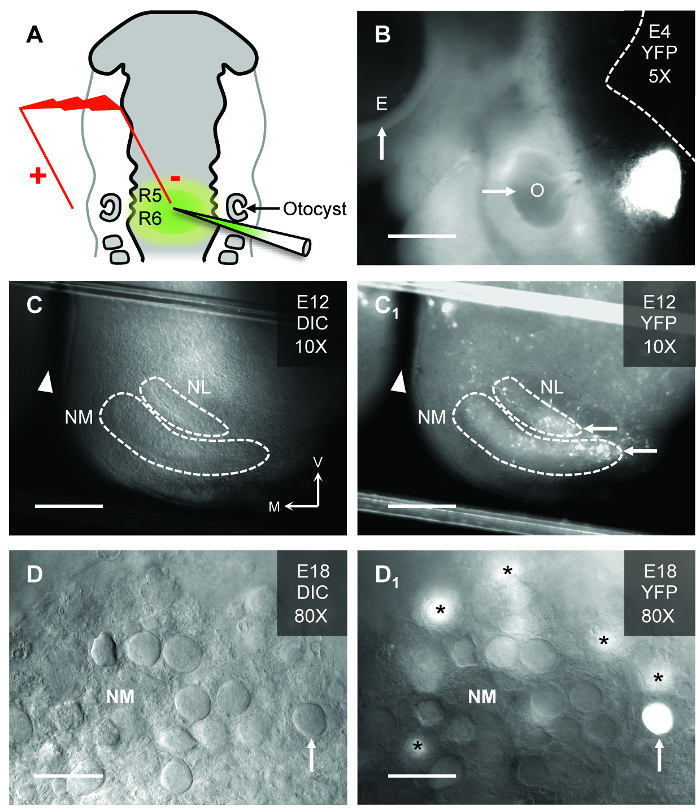
Figure 1: In Ovo Electroporation in the Chicken Auditory Brainstem. (A) Schematic of electroporation in the hindbrain of HH stage 12 chicken embryos. The injection pipette is filled with plasmid DNA and fast green dye for visualization of the injected hindbrain (rhombomeres 5/6 [R5/R6]). In this study, a plasmid that coexpressed a gene of interest and the yellow-fluorescent protein (YFP) reporter was used. Electroporated embryos are further incubated until desired developmental stage is reached. (B) Embryonic (E) day 4 chicken embryo showing site of YFP expression relative to the left eye (arrow, E) and left otocyst (arrow, O). Dashed white line represents the dorsal brain and brainstem border. Scale bar = 480 µm. (C and C1) Brainstem slice (300 µm thick) from an E12 chicken under differential interference contrast (DIC, C) and fluorescent (YFP, C1) illumination. Dashed white lines represent borders of nucleus magnocellularis (NM) and nucleus laminaris (NL). Note, the brainstem slice shows only one side of the tissue. White arrowheads in represents midline cleft of the brainstem slice. White arrows show YFP expressing regions of NM and NL. V = ventral, M = medial. Scale bar = 240 µm. (D and D1) High magnification (80X water immersion objective) of an E18 chicken embryo brainstem slice containing NM. The classic adendritic cell bodies15 are clearly visible under DIC illumination (upward arrow, D). With fluorescent illumination for the same slice, a transfected NM neuron identified by YFP fluorescence is clearly visible (upward arrow, D1). Asterisks = YFP expressing NM neurons just below the focal plane. Scale bar = 30 µm. Please click here to view a larger version of this figure.
Discussion
In ovo electroporation is a method of expressing or knocking down genes of interest in order to analyze in vivo gene function8,9. In the chicken embryo, it is an innovative method for expressing plasmid-encoded genes into different auditory brainstem regions8. To ensure optimal expression, several critical steps are required. First, only inject embryos whose otocysts are clearly visible. If otocysts are not visible the embryo is not at the correct developmental stage for electroporation. Second, smaller pipette tips make it easier to penetrate the neural tube and results in less tissue damage. However, the broken tip should be large enough to expel the DNA. Third, fill the entire neural tube region overlying R5/R6 with DNA (as visualized by spread of the fast green solution). Finally, placement of the stimulator electrode as close to the embryo as possible is important. However, make sure to avoid direct contact with the embryo, as current pulses can scorch and damage tissue.
Modifications to the technique can help improve the viability of embryos and the expression of plasmids. First, it is important to set eggs within 24 h of arrival to maintain high viability. Second, the window of the egg must be tightly sealed with tape as loss of fluid (i.e., dehydration) through an inadequate seal increases embryo death. Third, to optimize visualization of the otocyst, inject Indian ink from the rostral side of the embryo, using the smallest amount of ink as possible. Finally, stimulating electrodes can be moved slightly between R5/R6 regions to spread the electrically charged area without increasing current strength.
Despite the suggestions above, it should be noted that limitations exist. These include variable rates of transfected neurons, their location within the brainstem slice, and embryo survival. Although transfection efficiency varies amongst embryos, we previously reported with the electroporation parameter described here ~5-10% of NM/NL neurons are consistently transfected8. Even at higher rates of neuronal transfection, the preparation of brainstem tissue for in vitro patch-clamp electrophysiology poses challenges. For example, brainstem slices are between 200 – 300 µm thick and occasionally, transfected neurons are too deep within the tissue to adequately patch and perform functional assays. Finally, embryo survival post successful electroporation is also variable, albeit not as much as transfection rate. Depending on the desired embryo age for experimental use, survival variability can rage between 80-20%, the latter of which corresponds to the oldest embryos (>E18).
The significance of in ovo electroporation in chicken is advantageous over mammalian model systems for several reasons16. First, it is relatively inexpensive and an excellent alternative to transgenic or knockdown animals. Second, the chicken shares analogous nuclei with mammals that process temporal information of sound at the cellular, synaptic, and neural network level17. An example of similar auditory function occurs between AVCN/MSO in mammals and NM/NL of chickens, analogous auditory brainstem structures, respectively4,5. However, unlike traditional high-frequency-hearing mammalian research models, such as mice and rats, chickens utilize cues provided by both low- and high-frequency signals to encode auditory information18. Finally, chickens are auditory precocious animals; that is, their auditory system is near functional maturation at birth and the onset and refinement of hearing occurs during embryonic stages without environmental influences19,20. In contrast, the onset of hearing for typical mammalian research models begins 10-13 days after birth21,23.
In conjunction with biochemical, pharmacological, and functional assays, in ovo genetic manipulations provide an innovative approach to study neuron-specific development of anatomical and physiological specializations, permitting control of well-defined time periods in auditory brainstem development.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
We would like to thank Drs. Leslayann Schecterson, Yuan Wang, Andres Barria and Mrs. Ximena Optiz-Araya for initial assistance with protocol set-up and for providing plasmids. This work was supported by NIH/NIDCD grant DC013841 (JTS).
Materials
| Fertilized white leghorn chicken eggs | Sunnyside Inc. (Beaver Dam, WI) | ||
| Picospritzer | Parker Hannifin | 052-0500-900 | Picospritzer III, single or dual channel |
| Current/voltage stimulator | Grass Technologies | SD9 | SD9 |
| Microfil syringe needles | World Precision Instruments | MF28G67-5 | 28 Gauge, 67 mm Long, (Pack of 5) |
| Electrode holder | Warner Instruments | 64-1280 | MP Series: Non-Electrical Pressure Applications |
| Stimulating microelectrode | FHC | PBSA1075 | PBSA1075 |
| Air tank/regulator | NU Laboratory Services | Air dry 300 CF | |
| Fast green | Sigma Aldrich | F7258-25G | F7258-25G |
| Clear plastic tape | Scotch | 191 | |
| Doxycycline hyclate | Sigma Aldrich | D9891-1G | |
| Egg refrigerator | Vissani Wine Refrigerator | 13.3-16.1° C (56-61° F) | |
| Incubator | Hova-Bator | 37.8° C (100° F), ~50% humidity | |
| Dissection scope | Zeiss | 4.35E+15 | SteREO Discovery, V8 Microscope, 50.4X |
| Cold-light source | Zeiss | 4.36E+15 | CL6000 LED |
| Micromanipulators | Narishige Japan | Model: MM-3 | 2 Micromanipulators |
| Capillary tubes | Sutter Instrument | BF150-86-10 | Thick-walled borosilicate (dimensions) |
| Syringes | 1 mL, 3 mL | ||
| Needles | BD Precision Glide | 27 G x 1 1/4, 19 G x 1 1/2 | |
| Forceps | Stoelting | No. 5 Super Fine Dumont | |
| Egg holder | Custom Made | Clay base works as well | |
| Micropipette puller | Sutter Instrument | Model P-97 | |
| Syringe filter | Ultra Cruz | sc-358811 | PVDF 0.22 μm |
Referencias
- Grothe, B., Pecka, M., McAlpine, D. Mechanisms of sound localization in mammals. Physiol Rev. 90 (3), 983-1012 (2010).
- Anderson, S., et al. Neural timing is linked to speech perception in noise. J Neurosci. 30 (14), 4922-4926 (2010).
- Shannon, R. V., et al. Speech recognition with primarily temporal cues. Science. 270 (5234), 303-304 (1995).
- Carr, C. E., et al. Evolution and development of time coding systems. Curr Opin Neurobiol. 11 (6), 727-733 (2001).
- Carr, C. E., Soares, D. Evolutionary convergence and shared computational principles in the auditory system. Brain Behav Evol. 59 (5-6), 294-311 (2002).
- Rubel, E. W., Parks, T. N. Organization and development of brain stem auditory nuclei of the chicken: tonotopic organization of n. magnocellularis and n. laminaris. J Comp Neurol. 164 (4), 411-433 (1975).
- Rubel, E. W., Smith, D. J., Miller, L. C. Organization and development of brain stem auditory nuclei of the chicken: ontogeny of n. magnocellularis and n. laminaris. J Comp Neurol. 166 (4), 469-489 (1976).
- Schecterson, L. C., et al. TrkB downregulation is required for dendrite retraction in developing neurons of chicken nucleus magnocellularis. J Neurosci. 32 (40), 14000-14009 (2012).
- Chesnutt, C., Niswander, L. Plasmid-based short-hairpin RNA interference in the chicken embryo. Genesis. 39 (2), 73-78 (2004).
- Oertel, D. Encoding of timing in the brain stem auditory nuclei of vertebrates. Neuron. 19 (5), 959-962 (1997).
- Cramer, K. S., Fraser, S. E., Rubel, E. W. Embryonic origins of auditory brain-stem nuclei in the chick hindbrain. Dev Biol. 224 (2), 138-151 (2000).
- Cramer, K. S., et al. EphA4 signaling promotes axon segregation in the developing auditory system. Dev Biol. 269 (1), 26-35 (2004).
- Korn, M. J., Cramer, K. S. Windowing chicken eggs for developmental studies. J Vis Exp. (8), e306 (2007).
- Sanchez, J. T., et al. Preparation and culture of chicken auditory brainstem slices. J Vis Exp. (49), (2011).
- Jhaveri, S., Morest, D. K. Neuronal architecture in nucleus magnocellularis of the chicken auditory system with observations on nucleus laminaris: a light and electron microscope study. Neurociencias. 7 (4), 809-836 (1982).
- Matsui, R., Tanabe, Y., Watanabe, D. Avian adeno-associated virus vector efficiently transduces neurons in the embryonic and post-embryonic chicken brain. PLoS One. 7 (11), e48730 (2012).
- Koppl, C. Auditory nerve terminals in the cochlear nucleus magnocellularis: differences between low and high frequencies. J Comp Neurol. 339 (3), 438-446 (1994).
- Hyson, R. L. The analysis of interaural time differences in the chick brain stem. Physiol Behav. 86 (3), 297-305 (2005).
- Jones, T. A., Jones, S. M., Paggett, K. C. Emergence of hearing in the chicken embryo. J Neurophysiol. 96 (1), 128-141 (2006).
- Saunders, J. C., Coles, R. B., Gates, G. R. The development of auditory evoked responses in the cochlea and cochlear nuclei of the chick. Brain Res. 63, 59-74 (1973).
- Woolf, N. K., Ryan, A. F. The development of auditory function in the cochlea of the mongolian gerbil. Hear Res. 13 (3), 277-283 (1984).
- Walsh, E. J., McGee, J. Postnatal development of auditory nerve and cochlear nucleus neuronal responses in kittens. Hear Res. 28 (1), 97-116 (1987).
- Uziel, A., Romand, R., Marot, M. Development of cochlear potentials in rats. Audiology. 20 (2), 89-100 (1981).
