Most fluorescence studies inside living cells depend on protein fusions with fluorescent proteins (FPs), such as GFP1. These fluorescent tags allow studies of the copy number, diffusion pattern or localization of proteins involved in processes such as gene expression or membrane transport2-7. FPs offer high labeling specificity, easy implementation, and are available in a large inventory of variants with various photophysical and chemical properties1. However, organic fluorophores remain the prime choice for in vitro experiments due to their greater photostability (up to 100-fold more stable than FPs)8,9, small size (up to 100-fold smaller volume than FPs) and ease of intramolecular labeling (mainly through the use of cysteine residues). All these factors are particularly important for single-molecule fluorescence and FRET studies10.
Several internalization methods combining the advantages of organic labeling and in vivo detection have been introduced over the past decade; however, such methods either employ relatively large polypeptides tags (e.g., TMP, HALO, or 20 kDa SNAP tags)11-14, require the use of unnatural amino acids15, or are limited to large, single-membrane eukaryotic cells (e.g., scrape loading, syringe loading, microinjection)16-19.
This protocol describes a novel, straightforward and high-throughput internalization method that couples the advantages of organic fluorophores with in vivo observation. To develop this technique, we adapted the electroporation procedure commonly used to transform cells with plasmid DNA20,21 in order to load microorganisms, such as E. coli or S. cerevisiæ with organically labeled biomolecules. The protocol consists of 4 simple steps: incubation of cells with labeled biomolecules, electroporation, cell recovery, and cell washing to remove non-internalized biomolecules. Here, we present this electroporation protocol, as well as the cell imaging and data analysis processes to study cell-based and single-molecule fluorescence and FRET signals.
Electroporation relies on discharging a high-voltage electric field across a low ionic strength cell suspension to form transient membrane pores through which biomolecules can enter cells (Figure 1)20,21. Just as with transformation of bacteria or yeast with plasmid DNA, cells have to be prepared prior to electroporation to ensure their electrocompetency. This procedure, consisting of several washing steps with water, increases the membrane permeability and lowers the ionic strength of the cell solution to avoid arcing in the electroporation cuvette. In this protocol, cells can be prepared as described below (See PROTOCOL: 1.1) or bought from commercial providers.
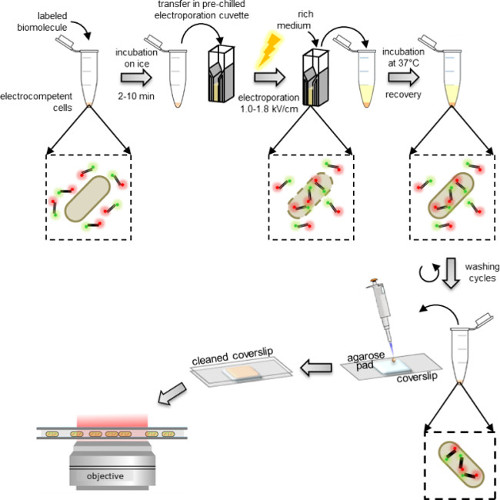
Figure 1: Schematic representation of the internalization protocol. From left to right: add a few microliters of labeled biomolecules to the aliquot of electrocompetent cells (doubly-labeled DNA fragments and bacteria in this example); incubate 1 to 10 min on ice and transfer to a pre-chilled electroporation cuvette; electroporate and then add 0.5-1 ml rich medium to the cells immediately after; incubate at 37 °C (or the temperature required by the organism, e.g., 29 °C for yeast) to let the cells recover; perform 5 washing steps to remove any excess non-internalized labeled molecules; resuspend the final pellet in 100-200 μl of PBS buffer and pipette 10 μl on an agarose pad; cover the pad with a cleaned coverslip and image on a fluorescence microscope (in wide field mode or HILO mode).
Electrocompetent cells are incubated with the labeled biomolecules just before electroporation, which can be performed using standard electroporators found in most biochemistry laboratories. Immediately after electroporation, cells are incubated in a rich medium allowing their recovery before washing (Figure 1). The excess of non-internalized labeled biomolecules is first removed by washing in a buffer containing a fairly high concentration of salt and some detergent (See PROTOCOL: 3.3). The presence of salt disrupts non-specific electrostatic interactions formed by non-internalized labeled biomolecules which otherwise may stick on the outer membrane. Similarly, the presence of detergent in the washing buffer disrupts non-specific hydrophobic interactions.
While DNA internalization is straightforward (Figure 2), precautions need to be taken when internalizing labeled proteins using electroporation. First, the stock sample of organically labeled protein might still contain a small percentage of free dye. Free dye molecules are much smaller than proteins and might therefore be internalized preferentially. To ensure that the vast majority of the observed internalized fluorescent molecules correspond to the protein of interest, the initial protein sample should contain less than ~2% free dye (Figure 5)22. The excess of non-internalized labeled proteins can also stick to the outer cell membrane after electroporation; this phenomenon is protein-specific and needs to be checked for each new protein. We propose several options that allow the removal of non-internalized proteins from the loaded cell sample (See PROTOCOL: 3.3.3).
Finally, cells are resuspended in a small volume of phosphate buffer and pipetted onto an agarose pad, allowing their imaging on a fluorescence microscope. Immobilization on agarose pads is a simple and efficient way of imaging cells on a coverslip without harming their integrity. The pad should contain a low-fluorescence culture medium.
Cell imaging can be performed either in widefield, total internal reflection fluorescence (TIRF) or using HILO (Highly Inclined and Laminated Optical Sheet) microscopy. In the HILO configuration, the laser beam penetrates deeper into the specimen than in TIRF, yet does not illuminate the entire sample as for widefield, allowing a greater signal-to-noise ratio23. Depending on the laser power and time resolution used, internalized biomolecules can be counted (using stepwise-photobleaching analysis, Figure 3), localized, or tracked24-28. Internalization of doubly labeled constructs with a FRET pair of fluorophores allows the quantification of FRET both at single-cell or single-molecule levels (Figure 6).
Different parameters can be varied depending on the desired output and the biological system studied. First, the amount of internalized material per cell can be tuned by altering the concentration of labeled biomolecules added to the cells prior electroporation (Figure 2). Electroporation field strength will also influence both the loading efficiency and cell viability; as expected, while the loading efficiency increases with increasing field strength, the viability of electroporated cells decreases (Figure 4A). Both parameters can be quantified by recording the percentage of loaded and dividing cells after electroporation. This viability assay coupled with fluorescence imaging also verifies the observation of internalized biomolecules in living cells and allow continuous observation over several generations (Figure 4B).
In summary, this protocol allows the internalization of fluorescently labeled DNA and protein molecules into E.coli or S. cerevisiæ26. Individual molecules labeled with organic fluorophores can be tracked with high spatiotemporal resolution for timescales an order of magnitude longer than FPs. Finally, this method is compatible with widefield, TIRF and confocal detection, as well as pulsed excitation schemes, such as ALEX (alternating laser excitation28,29).
Sample preparation
The different steps of the protocol are presented as schematics in Figure 1. As an example, we represented the loading of bacteria with doubly labeled (donor and acceptor dyes) DNA fragments. Representative results for DNA internalization are shown in Figure 2. For each electroporated sample, data for empty cells and non-electroporated cells were also recorded (Figure 2). “Empty cells” correspond to electrocompetent cells neither incubated with fluorescent biomolecules nor electroporated; their intensity in the fluorescent channel reflects the autofluorescence level under identical experimental conditions (laser power, time resolution, temperature, etc.). “Non-electroporated cells” (also called –EP, i.e., minus EP) correspond to a negative control in which electrocompetent cells have been incubated with the fluorescent biomolecules but not electroporated. These non-electroporated cells should exhibit a fluorescence level similar to the autofluorescence of the empty cells and significantly lower than the fluorescence intensity displayed by loaded, electroporated cells. This confirms the removal of any non-internalized labeled biomolecules that could have adhered to the outer cell membrane.
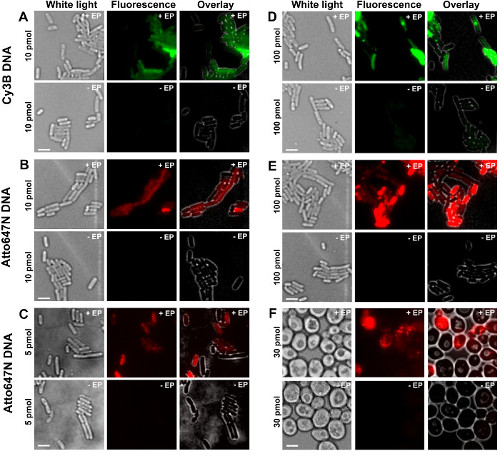
Figure 2: Representative results for the internalization of dsDNA labeled with different fluorophores at different concentrations in bacteria (A-E) and yeast (F). Left to right: white-light, fluorescence and overlay images. -/+ EP denotes incubation without/with electroporation. Scale bars: 3 µm. A. Cy3B dsDNA, 10 pmol, E. coli. B. ATTO647N dsDNA, 10 pmol, E. coli. C. Alexa647 dsDNA, 5 pmol, E. coli. D. Cy3B dsDNA, 100 pmol, E. coli. E. ATTO647N dsDNA, 100 pmol, E. coli. F. ATTO647N dsDNA, 30 pmol, Yeast. This figure has been modified from reference 26. Please click here to view a larger version of this figure.
Counting the number of internalized biomolecules per cell
The procedure to estimate the number of internalized labeled biomolecules per cell using photobleaching analysis is presented in Figure 3 and Supplementary Movie 2, together with representative results obtained with different concentrations of labeled DNA. Cell loading efficiency increases with the initial amount of incubated labeled DNA, allowing the user to tune the number of labeled molecules per cell from a “single-molecule” level (<10, Supplementary Movie 2B) to an “ensemble” level (>10, Supplementary Movie 2A). A robust way of estimating the percentage of loaded cells is to count the number of electroporated cells displaying a cell intensity above the average cell intensity of non-electroporated cells plus 3 times their standard deviation (i.e., Av(I–EP)+3*StdDev(I–EP) where Av = average, I = intensity per pixel, Std. Dev. = standard deviation and –EP = non-electroporated) as shown in Figure 3.
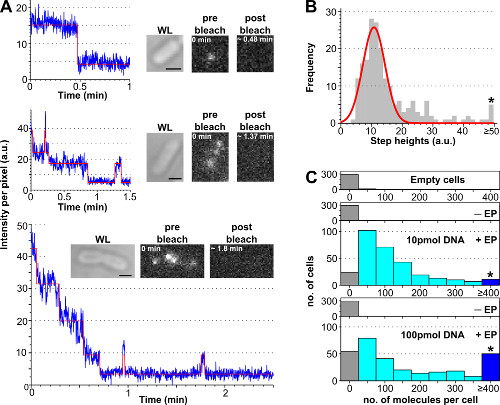
Figure 3: Counting the number of internalized molecules using photobleaching analysis. (A) Single-cell photobleaching analysis. Examples of fluorescence intensity timetraces (blue: raw data; red: fit; insets: WL and fluorescence images of E. coli loaded with ATTO647N-labeled dsDNA before and after bleaching). Top: single-step bleaching event. Middle: cell containing ±3 molecules showing bleaching and blinking. Bottom: cell containing >10 steps corresponding to at least 10 molecules. (B) Histogram of single-step height intensities from an automated step-fitting algorithm from 57 cells containing less than 6 distinguishable steps. Single-Gaussian fit is centered at 11 ± 3 a.u., corresponding to a unitary fluorophore intensity of 8100 photons per second. The asterisk marks the bin gathering all the step heights above or equal 50 a.u. (C) Histogram of internalized molecules per cell electroporated with different amounts of ATTO647N dsDNA, calculated after dividing the initial fluorescence intensity by the unitary fluorophore intensity. Top to bottom: empty cells (i.e., not incubated with fluorescent molecules and not electroporated), non-electroporated (but incubated with fluorescent molecules, named –EP), and electroporated cells incubated with 10 and 100 pmol dsDNA (named +EP). Empty and non-electroporated cells correspond to autofluorescence, whereas electroporated cells show a broad distribution of internalized molecules, with a higher proportion of highly loaded cells at 100 pmol (≥ 4 molecules, see asterisk-marked bin). Internalization efficiency (fraction of cells with Int. > mean + 3x Std. Dev. of non –EP sample) for the 10 and 100 pmol samples was 94% and 90%, respectively. Mean number of internalized molecules per cell: 121 ± 106 molecules for 10 pmol dsDNA, and 176 ± 187 molecules for 100 pmol dsDNA. Settings: 100 ms exposure, widefield illumination. Scale bars: 1 μm. This figure has been modified from reference 26. Please click here to view a larger version of this figure.
Cell loading and viability
In addition to changing the amount of labeled biomolecules added to the cells prior to electroporation, the user can tune the amount of internalized molecules by choosing different field strengths during electroporation (Figure 4, Supplementary Movie 1). Higher field strengths lead to greater internalization efficiencies, but lead to a slight decrease of cell viability. For protein internalization, the use of a filtration step may help remove non-internalized labeled proteins (see 3.3.3.1.1). In such cases, cell filtration ensures that observed fluorescent proteins are indeed internalized inside the bacterial cytoplasm; we note, however, that filtration also has a negative impact on cell viability (for more details, see REF 22).
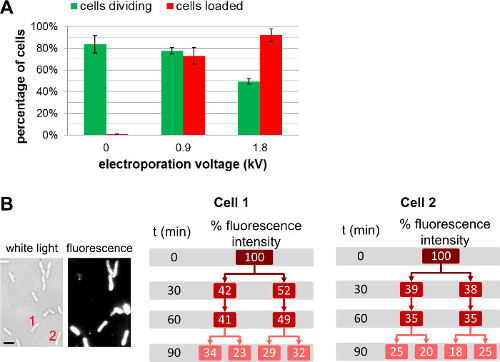
Figure 4: Influence of electroporation voltage upon cell loading and viability. (A) Bar chart representing the effect of electroporation field strength on loading efficiency (red bars) and viability of the cells (green bars). 84 ± 8 % of the non-electroporated cell (0 kV/cm) divide after 1 h on an agarose pad at 37 °C. Under the same conditions, 78 ± 3 % and 49 ± 3 % of the cells electroporated at 0.9 kV/cm and 1.8 kV/cm respectively divide after 1 hr. For loading efficiency, 73 ± 8 % of the cells are loaded at 0.9 kV/cm, while 92 ± 6 % of the cells are loaded at 1.8 kV/cm. The error bars represent the standard deviation calculated from three independent measurements; more than 200 cells were analyzed for each sample and each repeat. Overall loading efficiency increases with electroporation voltage to the slight detriment of cell viability. (B) Cell-based fluorescence measurements over several generations show that the overall fluorescence intensity is shared equally between both daughter cells. Cell 1 and 2 refers to the cell number in the white light image (left) and fluorescence image at t = 0. Scale bar: 1 µm). This figure has been modified from reference 26.
Protein internalization
Representative results for protein internalization are in Figures 5A & B. It is particularly important to remove as much of the remaining free (unreacted) dye from the protein sample as possible prior to electroporation. In the examples in Figures 5A & B, the Cy3b-labeled Klenow Fragment sample (Cy3b-KF, where KF is the Klenow fragment of the E. coli DNA Polymerase I, 66 kDa) only contains 1% free dye; such dye contribution to the overall cell loading is negligible. Comparisons of the electroporated sample of interest with both the non-electroporated cells (incubated with the same amount of labeled protein) as well as cells electroporated with the equivalent amount of free dye constitute two required controls to ensure that the observed fluorescent molecules are indeed internalized labeled proteins.
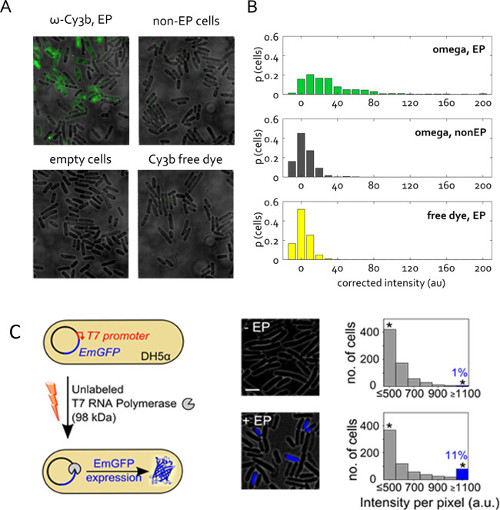
Figure 5: Protein internalization in live bacteria. (A) Representative fluorescence overlay field of views. Cells electroporated at 1.4 kV voltage with 50 pmol RNAP ω subunit from a protein stock solution that contained only 1% free Cy3b dye. Non-electroporated (non –EP) and empty cells are defined as previously. Free dye was internalized at the same concentration as in the RNAP ω electroporated sample. Imaging in wide field mode, 532-nm excitation at 1 mW, 50 ms exposure. (B) Distribution of uncorrected cell-averaged intensities for samples in (A), given in proportion of the total cell count. More than 400 cells per sample were segmented. This figure has been modified from reference 22. (C) Internalization of unlabeled T7 RNA polymerase (T7 RNAP, 98 kDa) into electrocompetent DH5α carrying the pRSET-EmGFP plasmid encoding emerald GFP (EmGFP) under control of a T7 promoter. Left: Schematic of assay. Middle: fluorescence overlay. Right: histograms of cell-based fluorescence intensities for the non-electroporated sample (top) and cells incubated and electroporated with T7 RNAP (bottom); approximately 11% of the electroporated cells show high fluorescence intensity (fraction of cells with Int. > mean + 3x Std. Dev. of non –EP sample) indicating expression of EmGFP. The asterisks indicate bins gathering all the intensities above or equal 1,100 a.u. Scale bar: 3 μm. This figure has been modified from reference 26.
Figure 5C presents another application of protein electroporation. Here, the electroporated protein is unlabeled, but its internalization triggers an observable fluorescent response. This experiment verifies the presence and functionality of electroporated proteins in the cell cytoplasm. Unlabeled T7 RNA polymerase (98 kDa) was internalized into E. coli strain DH5α containing a plasmid encoding for fluorescent protein EmGFP under the control of a T7 promoter26. As the gene for T7 RNA polymerase is absent in DH5α, EmGFP expression in our experiments requires that functional T7 RNA polymerase is introduced into the cells via electroporation (Figure 5C). Following electroporation with 1 pmol T7 RNAP, >11% of the cells (blue bar, Figure 5C) exhibited fluorescence higher than the negative control (incubated with the same amount of T7 RNAP, but not electroporated). This result establishes that a proportion of the T7 RNAP molecules internalized by electroporation retain their integrity in vivo and can perform their intended functions in the cell cytoplasm.
In vivo FRET at the single-molecule and single-cell levels
Finally, the internalization and analysis of doubly labeled species in living bacteria is presented in Figure 6 and Supplementary Movie 3. As fluorescent protein fusions are not ideal for in vivo smFRET studies, the ability to deliver doubly labeled biomolecules into living cells using electroporation is one of the great assets of this method. Figure 6A presents single-cell FRET analysis of bacteria loaded with different FRET DNA standards (using Cy3B and Atto647N fluorophores as donor-acceptor FRET pair). Cells are electroporated with 20 pmol of three short doubly-labeled dsDNA FRET standards with apparent FRET efficiencies (E*) of 0.17, 0.48, and 0.86 in vitro (previously determined26). All DNAs enter cells efficiently (Figure 6A, left) and the main peak of each single-cell E* distribution agrees well with the in vitro results (Figure 6A, right). In the intermediate- and high-FRET samples, cell populations with lower E* than expected are observed, presumably due to a combination of acceptor bleaching and photophysical inactivity, variable cell loading (thus, variable signal-to-noise ratio) and DNA degradation.
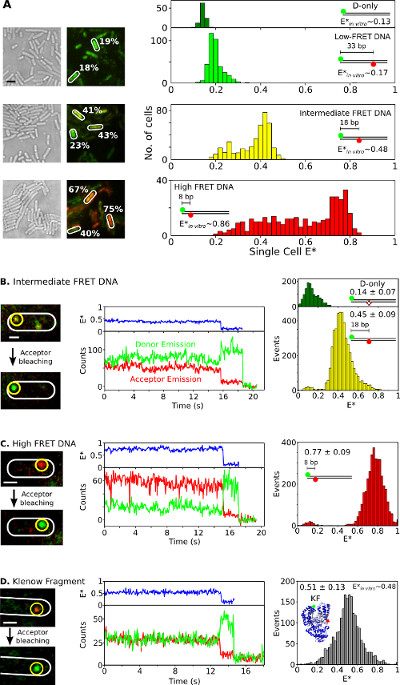
Figure 6: Representative results for single-cell and single-molecule FRET observation in living bacteria. Ensemble and smFRET studies in single bacteria. (A) Analysis of cells loaded with 20 pmol each of three DNA FRET standards exhibiting low (~0.17), intermediate (~0.48), and high (~0.86) FRET (as measured using in vitro single-molecule measurements; see REF 26). Left: white light and green/red (FRET) fluorescence overlay images (Scale bar: 3 μm). Examples of FRET values from different cells are indicated (white). Right (top to bottom): uncorrected cell-based FRET (E*) histograms for donor only (dark green), low (light green), intermediate (yellow), and high (red) FRET DNA standards. (B–D) In vivo smFRET. Cells are loaded with 0.25 pmol intermediate-FRET DNA (panel B), 0.25 pmol high-FRET DNA (panel C), and 5 pmol doubly-labeled KF (panel D). Left column: green/red fluorescence overlay of single frame before and after acceptor photobleaching. Middle column: time traces corresponding to the molecule in the yellow circle. FRET efficiencies, donor emission intensities, and acceptor emission intensities are displayed in blue, green, and red, respectively. Right column: FRET histograms of donor only molecules (green) and donor-acceptor molecules (yellow, red, and gray) from 20 time traces for each sample. Scale bars: 3 μm for A, 1 μm for B–D. This figure has been modified from reference 26. Please click here to view a larger version of this figure.
To observe smFRET in vivo for DNA or protein samples, low amounts (0.25 pmol) of intermediate- and high-FRET DNA standards (Figure 6B, C) or 5 pmol of double-labeled KF (Alexa647/Cy3B fluorophores as FRET pair, Figure 6D) are electroporated into E. coli. Such concentrations led to many cells loaded with few (n = 1–10) labeled molecules, allowing direct localization, tracking, and FRET monitoring for single molecules. Some molecules diffuse freely, whereas others appear immobile or diffuse slowly (Supplementary Movie 3). Timetraces of immobilized doubly-labeled biomolecules (Figure 6, middle) last for 1 to 30 s and show the hallmarks of smFRET: anticorrelated changes in the donor and acceptor fluorescence upon acceptor bleaching (for example, t~16 sec; Figure 6B, middle), followed by single-step donor bleaching (for example, t~;19 sec; Figure 6B). FRET distributions generated from such timetraces (Figure 6, right) result in a mean value that is in excellent agreement with published in vitro studies26,31,32. These results establish the capability for quantitative smFRET studies on internalized DNAs and proteins, and suggest that proteins maintain their integrity and structure upon electroporation and internalization (as supported by the T7 RNAP internalization experiments).
Supplementary Movie 1: Cell viability. Left: White light images. Right: fluorescence images. Animated GIF animated showing the division after electroporation (1.8 kV/cm) of bacteria loaded with 10 pmol Atto647 labeled DNA. The overall apparent decrease of fluorescence is due to the dilution of labeled DNA upon cell division and also partially to the photobleaching which occurs during each measurement.
Supplementary Movie 2: Cell-based photobleaching studies. A. Representative example of a heavily loaded cell (containing >100 Atto647N-labeled DNA molecules). Top left, white light image of the cell of interest (red rectangle). Top right, movie of the loaded cells showing their fluorescence decay over several minutes. Bottom, time trace of the overall fluorescence intensity decay of the cell of interest. Organic fluorophores can exhibit photobleaching lifetimes 2 orders of magnitude higher than FPs (here, ~41 sec for Atto647N). B. Representative example of a cell loaded with less than 10 labeled molecules (3 in this case). Top, same as panel A. Bottom, time trace of the overall fluorescence intensity of the cell of interest showing single step bleaching and/or blinking corresponding to single organic fluorophores. The average height of these steps corresponds to the single-molecule unitary intensity (here ~12 a.u.) used to estimate the initial number of internalized molecules per cell. Movies under continuous red laser excitation at 300 µW power and 100 msec per frame.
Supplementary Movie 3: In vivo single-molecule FRET. Top: Cells loaded with 0.25 pmol high-FRET DNA (as in Figure 6C) continuously monitored at 50 ms per frame under nTIRF illumination using 1 mW green (532 nm) laser. Each frame is a green/red (FRET) fluorescence overlay of each channel. Diffusing and immobile red (intact) and green (single active label) DNA molecules can be observed. Bottom: Time trace corresponding to the molecule in the yellow circle. FRET efficiencies, donor emission intensities, and acceptor emission intensities are displayed in blue, green, and red, respectively. Anti-correlated acceptor bleaching event (red-to-green transitions) corresponds to the signature of single-molecule FRET.
