1. Selection of disease that may be caused by de novo mutations
A disease that corresponds to the following criteria can fit with the de novo mutation hypothesis:
- The reproductive fitness is reduced.
- The frequency of the disease is relatively high and constant despite widely varying environments.
- The disease is associated with a higher paternal age.
- The classic linkage and association studies failed to explain a significant fraction of the disease heritability.
- The twin concordance data support a de novo model.
Analysis of the likelihood that a common disease where de novo mutations may in part explain the genetic basis is a critical first step.
2. Selection of cases and DNA samples
Selection of appropriate samples is critical for the success of the identification of de novo mutations. To maximize the chance of finding de novo mutations, we recommend the following:
- Select cases with early age of onset, severe phenotype, with unaffected parents, older fathers and with no extended family history of the disease.
- Choose patients whose available DNAs are sufficient to conduct the study. Especially critical is the availability of DNA from a primary cell source that was not subjected to culturing (e.g. Blood DNA or saliva DNA),
- The availability of both parents DNA is critical in order to determine the mutation transmission status (inherited vs. de novo). Availability of additional affected cohorts and normal controls is necessary for genetic validation studies once candidate genes are identified.
- Estimate the sample size based on the mutations rate, the amount of genes to be screened and the estimate of the fraction of cases that may result from a de novo mutation.
3. Gene resequencing; two major approaches
- High quality low throughput sequencing
This approach is based on the candidate genes approaches.- Selection of candidate gene(s)
Select the best candidate genes based on a scoring system which is built on 6 major criteria. Then calculate the total that corresponded to the sum of all the points attributed using the six criteria listed in Table 1. See example from our project in figure 1 of selected and not selected genes distribution.

Table 1. Criteria used for the candidate gene selection
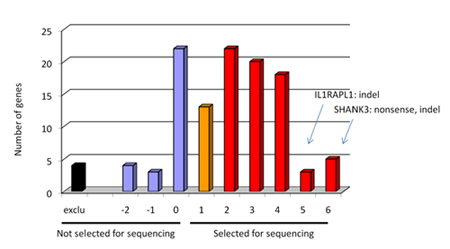
Figure 1. Graph showing the distribution of selected and not selected genes for sequencing in our project. We obtained a distribution of genes ranked by candidate properties by sorting genes according to their score value. For example, SHANK3 and NRXN1 genes, two genes that we found de novo mutation, had a score 7 and 6 respectively (maximum is 12). - Design primers using Primers3 software through Exonprimer. Only coding region and splice junction should be covered including an extra 50 base pairs on each side of the exon.
- Optimize PCR conditions for the choice of Taq, reaction volume, etc.
- Optimize all PCR fragments
- Amplify 5 ng of genomic DNA extracted from blood samples according to standard procedures
- Before sending for sequencing, do quality control of your PCR products by loading a 2% agarose gel. Selected randomly samples.
- Sequence the PCR products on a DNA Analyzer on one strand. A fragment is considered successfully sequenced if the analysis of over 90% of the traces is possible. This is applicable for a large scale screening.
- Variants Detection
- Use tools for detection and genotyping of genomic variations such as PolyPhred, Polyscan and Mutation Surveyor. A combination of more than 2 detection tools is ideal. For example, PolyPhred v.5 and PolyPhred v.6 with the default settings do not detect the same variations. Polyscan v.3 has a higher false positive mutation rate for SNPs (96%) and less for the INDELs (93%). PolyPhred v.6 did not detected the majority of true INDELs but have a false positive mutation rate (for INDELs) lower than Polyscan v.3 overall (90%). We should remove the option of SNPs detection for Polyscan v.3 and keep both PolyPhred v.5 and PolyPhred v.6 for variant detection. Mutation Surveyor and Polyscan are better for detecting indels. Note: The option of SNP detection should not be applied when using Polyscan. Only the “indel” option should be on. Polyscan generated to many false positive for SNP detection.
- For each unique novel exonic variants detected, confirm it manually by reamplifying the fragment and resequencing the proband and both parents using reverse and forward primers to eliminate any technical artifact.
- Selection of candidate gene(s)
- Whole exome sequencing
This approach is a high throughput sequencing targeting the majority of coding regions of the human genomes. We are now currently using this new approach in our lab, accelerating the detection of potential candidate genes.- Order the “SureSelect Human All Exon” targeting the coding region of over 16,000 genes (50 MB of the genome) designed by Agilent or any other similar product by others like Roche . Prior to order the capture kit, the sequencing platforms should be determine (Illumina vs SOLiD).
- Do the capture according to the Agilent protocol
- Sequence the product on your respective available next-generation platform
- Variant detection from the whole exome sequencing: Several bioinformatics tools for detection and genotyping of genomic variations from the next-generation sequencing platform are available such as BWA, Bfast, Bioscope which will perform the alignment. After which additional freely available downstream tools (for example SAM tools, Varscan, Annovar) would be needed it to call and annotate the variants. Commercial software that incorporates sequence alignment and variant calling and annotation are also available such as, NextGEN (Softgenetics), CLC Bio, and others
4. Genomic variants prioritization
Identified variants are then prioritized for follow up according to their probability in being de novo and deleterious to protein or mRNA function and /or structure. The variant follow up priorities for detection of de novo variant should be as follow:
- Unique variations (observed once in a single case)
- Variations not present in the parents
- Protein-truncating variations: nonsense, indels leading to frameshift and splicing mutations.
- Missense and silent variation predicted to be functionally disruptive (e.g., affect mRNA splicing). Use Polyphen, SIFT and PANTHER for functional prediction effect on the protein.
If using whole exome sequencing, selection of candidate genes can be used as a strategy for prioritizing variants for further study.
5. Genetic validation
- Resequence the entire gene in additional patient cases (to identify other causative mutations) and in controls. The control samples will be used to evaluate allelic frequencies of prioritized variants. Any gene containing at least two different de novo deleterious mutations (nonsense, splicing, frameshifts, and predicted damaging missense) found in different patients (but not in control samples or public databases) should be highly prioritized for further validation studies. This includes: 1) testing for a potential splicing defect in lymphoblastoid cell lines derived from the patient. In our experience, most genes yield RT-PCR products from lymphoblastoid cell lines; 2) investigate altered protein expression levels by quantitative Western blot analysis of protein extracts from the lymphoblastoid cell lines and 3) further test the mutation/gene at the functional level in animal (c elegans, Zebrafish) and cell line models as previously done by our group (ex: 5,6).
6. Representative Results:
Following this protocol, we were able to identify new genes for schizophrenia and autism. One example is our recently SHANK3 gene discovery (Figure 2). Two different de novo mutations in SHANK3 gene, one nonsense mutation found in three affected brother and one missense mutation in one affected female.
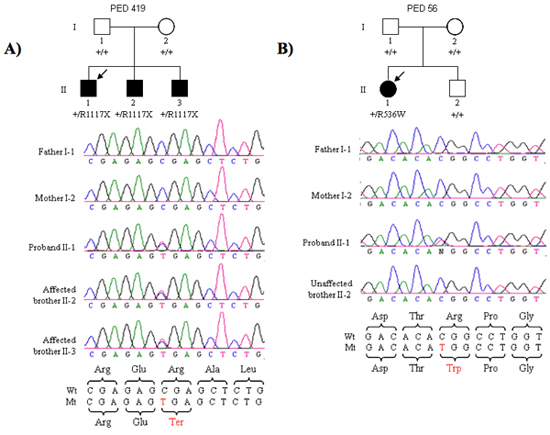
Figure 2. (A) Segregation of the R1117X nonsense mutation in three affected brothers of family PED 419. The proband is indicated by the arrow. (B) Segregation of the R536W missense mutation in the proband but not her non-affected brother in PED 56.
