Determining Intestinal Permeability Using Lucifer Yellow in an Apical-Out Enteroid Model
Summary
The present protocol outlines a method that utilizes lucifer yellow in an apical-out enteroid model to determine intestinal permeability. This method can be used to determine paracellular permeability in enteroids that model inflammatory bowel diseases such as necrotizing enterocolitis.
Abstract
Enteroids are an emerging research tool in the study of inflammatory bowel diseases such as necrotizing enterocolitis (NEC). They are traditionally grown in the basolateral-out (BO) conformation, where the apical surface of the epithelial cell faces the inner lumen. In this model, access to the luminal surface of enteroids for treatment and experimentation is challenging, which limits the ability to study host-pathogen interactions. To circumvent this, a neonatal apical-out (AO) model for necrotizing enterocolitis was created. Since intestinal epithelial cell permeability changes are pathognomonic for NEC, this protocol outlines using lucifer yellow (LY) as a marker of paracellular permeability. LY traverses the intestinal epithelial barrier via all three major paracellular pathways: pore, leak, and unrestricted. Using LY in an AO model allows for a broader study of permeability in NEC. Following IRB approval and parental consent, surgical samples of intestinal tissue were collected from human preterm neonates. Intestinal stem cells were harvested via crypt isolation and used to grow enteroids. Enteroids were grown to maturity and then transformed AO or left in BO conformation. These were either not treated (control) or were treated with lipopolysaccharide (LPS) and subjected to hypoxic conditions for the induction of in vitro NEC. LY was used to assess for permeability. Immunofluorescent staining of the apical protein zonula occludens-1 and basolateral protein β-catenin confirmed AO conformation. Both AO and BO enteroids treated with LPS and hypoxia demonstrated significantly increased paracellular permeability compared to controls. Both AO and BO enteroids showed increased uptake of LY into the lumen of the treated enteroids compared to controls. The utilization of LY in an AO enteroid model allows for the investigation of all three major pathways of paracellular permeability. It additionally allows for the investigation of host-pathogen interactions and how this may affect permeability compared to the BO enteroid model.
Introduction
Enteroids are three-dimensional (3D) structures derived from organ-restricted human intestinal stem cells1,2. They are made up entirely of epithelial lineage and contain all the differentiated intestinal epithelial cell types2. Enteroids also maintain cellular polarity made up of an apical luminal surface forming an inner compartment and a basolateral surface facing the surrounding media. Enteroids are a unique model in that they preserve the characteristics of the host from which they were generated3. Thus, enteroids generated from premature human infants represent a model that is useful for investigating diseases that primarily affect this population, such as necrotizing enterocolitis (NEC).
The traditional enteroid model is grown in a basolateral-out (BO) conformation, where the enteroid is encased in a dome of basement membrane matrix (BMM). BMM induces the enteroid to maintain a 3D structure with the basolateral surface on the outside. BO enteroids are a suitable model for NEC that bridges the gap between two-dimensional (2D) primary human cell lines and in vivo animal models2,4. NEC is induced in enteroids by placing pathogens such as LPS or bacteria in the media surrounding the enteroids, followed by exposure to hypoxic conditions2,3. The challenge with the BO enteroid NEC model is that it does not allow for the effective study of host-pathogen interactions, which occur at the apical surface in vivo. Changes in intestinal permeability are due to these host-pathogen interactions. To better understand how permeability affects the pathophysiologic basis of disease, a model must be created that involves treating the apical surface.
Co et al. were the first to demonstrate that mature BO enteroids can be induced to form an apical-out (AO) conformation by removing the BMM domes and resuspending them in media5. This article demonstrated that AO enteroids maintained correct epithelial polarity, contained all intestinal cell types, upheld the intestinal epithelial barrier, and allowed access to the apical surface5. Using AO enteroids as an NEC model achieves a physiological reproduction of the disease process and study of host-pathogen interactions.
One major contributor to the pathophysiology of NEC is increased intestinal permeability6. Several molecules have been proposed as a way to test for intestinal permeability in vitro7. Among these, lucifer yellow (LY) is a hydrophilic dye with excitation and emission peaks at 428 nm and 540 nm, respectively8. As it crosses through all the major paracellular pathways, it has been used to evaluate paracellular permeability in various applications, including the blood-brain and intestinal epithelial barriers8,9. The traditional application of LY uses cells grown in monolayers on a semi-permeable surface10. LY is applied to the apical surface and crosses through paracellular tight junction proteins to congregate on the basolateral side. Higher LY concentrations in the basolateral compartment indicate decreased tight junction proteins with subsequent intestinal epithelial cell barrier breakdown and increased permeability10. It has also been described in 3D BO enteroid models where LY was added to the media and individual enteroids were imaged for uptake of LY into the lumen11. Although this allows for qualitative analysis via the visualization of LY uptake, quantitative analysis is limited. This protocol outlines a unique technique that uses LY to assess paracellular permeability using an in vitro NEC enteroid model in AO enteroids while maintaining 3D orientation. This method can be used for both qualitative and quantitative analysis of permeability.
Protocol
The present research was performed in compliance with Institutional Review Board approval (IRB, #11610, 11611) at the University of Oklahoma. Parental consent was required prior to collecting human surgical specimens as per IRB specifications. Following IRB approval and parental consent, human small intestinal tissue was obtained from infants (corrected gestational age (GA) ranging from 36-41 weeks at the time of sample collection, all with a history of preterm birth at an estimated GA of 25-34 weeks, 2:1 M:F) undergoing surgery for NEC or other intestinal resection, such as ostomy takedown or atresia repair. Enteroids were generated from tissue obtained from either the jejunum or ileum.
1. Human infant-derived enteroid cultures: crypt isolation and plating from whole tissue
- Prepare culture media, chelating buffer #1, and chelating buffer #2 (Table 1) following the previous report4. Store chelating buffers at 4 °C and use them within 48 h.
- Prepare human minigut media (Table 1). Store the stock at −20 °C and warm in a 37 °C water bath prior to use.
- Prepare 50% L-WRN conditioned media (CM) (Table 1). Store the stock at 4 °C for up to 1 week.
NOTE: 100% L-WRN base for the conditioned media is described in a protocol by Miyoshi et al.12. - Generate enteroids from small intestinal tissue samples obtained from surgical specimens following the previous report4. Plate four wells of enteroids in a 24-well plate from a 0.75-2.5 g piece of intestinal tissue.
NOTE: Enteroids can be successfully generated from tissue stored in 30 mL Roswell Park Memorial Institute (RPMI) 1640 medium in a 50 mL conical tube at 4 °C for up to 48 h. - Place the 24-well plate in a 37 °C, 5% CO2 incubator upside-down for 15-20 min to allow for the polymerization of BMM domes4.
- Add 500 µL of 50% L-WRN CM (as described in step 1.3) with 0.5 µL of Y-27632, ROCK inhibitor (RI, see Table of Materials) to each well. After 2-3 days, replace with 50% L-WRN CM without RI.
2. Generation of AO enteroids
- Grow enteroids embedded in BMM for 7-10 days with 50% L-WRN CM4.
- Remove the media and add 500 µL of cell recovery solution (CRS, see Table of Materials) to one well. Scrape the dome, pipette up and down several times, then add the CRS/enteroid/BMM solution to the next well.
- Scrape the dome, pipette up and down several times, and place the solution in a microcentrifuge tube. Add 500 µL of CRS to the second well, pipette up and down, then add it to the first well. Transfer this solution to the same 1.5 mL microcentrifuge tube.
- Repeat step 2.3, pooling two wells into one microcentrifuge tube until all the well contents are in CRS.
NOTE: More than two wells can be pooled into one larger 15 mL conical tube if generating large volumes of AO enteroids. Maintaining a 500 µL CRS per well ratio is important to ensure complete solubilization of BMM. - Place the microcentrifuge tubes (step 2.3) with pooled CRS/enteroid/BMM solution on a rotator for 1 h at 4 °C to solubilize the BMM.
- Centrifuge the solution at 200 x g for 3 min at 4°C, then remove supernatant with a micropipette, leaving the pellet behind.
- Resuspend the enteroid pellet in 1 mL of 50% L-WRN CM (as prepared in step 1.3) per microcentrifuge tube. Gently pipette 500 µL of the re-suspended enteroid/media solution into each well of the ultra-low attachment 24-well tissue culture plates (see Table of Materials).
- Incubate at 37 °C with 5% CO2 for 3 days prior to experimentation.
3. Verification of AO enteroid conformation via whole-mount immunofluorescent staining
- Following incubation of the enteroids in media suspension for 3 days, pipette the enteroid/media suspension from one well into a microcentrifuge tube.
- Centrifuge the enteroid/media suspension at 200 x g for 5 min at 4 °C. Remove the supernatant with a micropipette.
- Resuspend the enteroids in 20 µL of BMM. Pipette 10 µL of enteroid suspension onto a microscope slide and spread it in a thin layer approximately 1 cm by 1 cm square. Repeat with the remaining 10 µL of enteroid suspension on a separate part of the same slide so that there are two smears of enteroid/BMM suspension.
- Allow the enteroid/BMM smears to solidify at room temperature (RT) for 15 min.
- Working in the fume hood, place the slide in a staining jar and fill with 4% paraformaldehyde until it covers the enteroid/BMM smear (~30 mL for one slide if using a glass staining jar with 10 slide capacity). Allow 30 min (at room temperature) for fixation to take place.
CAUTION: 4% paraformaldehyde is hazardous and may cause eye damage/irritation, skin irritation, and cancer. Always work under a fume hood when using it. Wear protective gloves and personal protective equipment when handling it. Dispose of it in an approved waste disposal container. - Discard 4% paraformaldehyde in an appropriate waste container. Add 30 mL of phosphate-buffered saline (PBS) to the staining jar or until PBS covers the enteroid/BMM smears. Let it sit for 3 min at RT, and then discard the PBS in the paraformaldehyde waste bottle. Repeat this step two more times for a total of three washes.
- Fill a new staining jar with 30 mL of 0.5% Triton X-100 diluted in PBS. Place the slide with enteroid/BMM smears in the Triton solution and let it permeabilize for 20 min at RT.
- Discard the 0.5% Triton X-100 diluted in PBS. Add 30 mL of PBS to the staining jar and let it sit for 3 min. Discard the PBS by decanting.
- Gently wipe the slide around the enteroid/BMM smears, being careful not to disrupt them. Using a hydrophobic barrier pen (barrier PAP pen, see Table of Materials), draw a circle around each of the enteroid/BMM smears. Make a 20% serum, specific to the animal in which the secondary antibody was raised (see Table of Materials).
- Lay the microscope slide flat on the lab bench. Block the slide for 1 h at 4 °C by pipetting 100 µL of specific 20% serum from step 3.9 onto each of the enteroid/BMM smears ringed by the hydrophobic barrier pen.
- Prepare a 100 µL solution with 1:100 and 1:200 dilutions of β-catenin and ZO-1 primary antibodies, respectively, diluted in PBS.
NOTE: Each primary antibody must be from a different animal to ensure that they will have distinct immunofluorescent channels when imaged. The present study utilizes a mouse β-catenin antibody and a rabbit ZO-1 antibody (see Table of Materials). - Tap the slide on the counter to remove the 20% serum and gently wipe dry, being careful not to disrupt the enteroid/BMM smears. Pipette 100 µL of β-catenin/ZO-1 primary antibody solution onto one of the enteroid/BMM smears. Pipette 100 µL of PBS without primary antibody onto the other enteroid/BMM smear as a negative control.
- Line the bottom of a plastic container with a wet paper towel to create a humidified container. Place the slide in the container, being careful not to disrupt solutions covering the enteroid/BMM smears. Place the lid on the container to seal, and store flat at 4 °C overnight.
NOTE: Do not allow the slide to dry out. Ensure that the container is closed to avoid desiccation. - Once ready to proceed, tap the slide on the counter to remove primary antibodies and PBS from the enteroid/BMM smears.
- Perform a wash step by placing the slide in a staining jar and filling with 30 mL of PBS or until PBS covers the enteroid/BMM smears. Let it sit for 3 min at room temperature. Discard the PBS and repeat this step twice more for a total of three washes.
- Prepare 200 µL of secondary antibodies for two different immunofluorescent channels, with each antibody having a 1:1000 dilution in PBS (see Table of Materials). Keep the solution away from light.
- Pipette 100 µL of secondary antibody solution onto each of the enteroid/BMM smears. Place it in the dark and let it sit for 1 h at RT.
- Tap the slide on the counter to remove secondary antibodies. Repeat the wash steps outlined in step 3.15, ensuring that all washes are performed in the dark.
- Add a drop of 4′,6-diamidino-2-phenylindole (Fluoroshield with DAPI, see Table of Materials) mounting medium to each of the enteroid/BMM smears and apply a coverslip to ensure that both smears are adequately covered. Avoid trapping bubbles under the coverslip. Keep the slide in the dark until ready to view under the microscope.
NOTE: Immediate imaging of the slides yields the most optimal results; however, slides can be stored flat in a humidified container at 4 °C and imaged for up to 1 week after the addition of DAPI.
4. Induction of experimental NEC
- Ensure that AO enteroids have been in suspension for at least 72 h prior to use.
- Gently pipette all the enteroid/media suspension from each well into a microcentrifuge tube.
NOTE: Multiple wells can be pooled into one 15 mL conical tube if a large volume of wells is treated. A minimum of three wells per treatment group is recommended for this protocol. - Centrifuge at 300 x g for 3 min at RT and remove the supernatant.
- Add 10 µL of 5 mg/mL of lipopolysaccharide (LPS, see Table of Materials) to 500 µL of 50% LWRN CM per well (final concentration 100 µg/mL LPS).
- Resuspend AO enteroids designated as the treatment group in 50% LWRN + LPS and aliquot 500 µL of suspension to a 24-well ultra-low attachment plate. Resuspend AO enteroids designated as untreated controls in 500 µL of 50% LWRN CM per well. Aliquot 500 µL of this suspension to a separate 24-well ultra-low attachment plate.
- Induce hypoxia in the LPS-treated AO enteroids via a modular incubator chamber (MIC) with 1% O2, 5% CO2, and 94% N2 as per the manufacturer's instructions (see Table of Materials). Treat the enteroids with hypoxia and LPS for 24 h.
- Place the untreated and LPS + hypoxia-treated cultures, still in the MIC chamber, in a 37 °C, 5% CO2 incubator for 24 h.
5. Measurement of paracellular permeability utilizing LY
- Gently pipette the enteroid/media suspension from each well into a microcentrifuge tube. Warm DPBS and 50% LRWN CM in a 37 °C water bath.
- Centrifuge the enteroid/media suspension at 300 x g for 3 min at RT.
- Remove the media. Wash the enteroids with warm 500 µL DPBS.
- Centrifuge at 300 x g for 3 min at RT. Remove the supernatant with a micropipette.
- Repeat steps 5.3-5.4 once more for a total of two times.
- After washing, add 450 µL of warm 50% LWRN CM (as prepared in step 1.3).
- Add 50 µL of 2.5 mg/mL LY (final concentration is 0.25 µg/µL, see Table of Materials) to each well and gently swirl to mix. Place in a 37 °C, 5% CO2 incubator for 2 h.
- Gently pipette the enteroid/media suspension from each well into a microcentrifuge tube.
- Centrifuge at 300 x g for 3 min at RT, then remove the supernatant, leaving the enteroid pellet behind.
- Wash the enteroids with 500 µL warm DPBS.
- Centrifuge at 300 x g for 3 min at RT, then remove the supernatant leaving the enteroid pellet behind.
- Repeat steps 5.10-5.11 three more times for a total of four washes.
- Resuspend each pellet with 1,000 µL of cold DPBS and vigorously pipette up and down to dissociate the enteroids.
- Prepare standards for the LY standard curve diluted in DPBS (100 ng/mL; 50 ng/mL; 25 ng/mL; 12.5 ng/mL; 6.25 ng/mL; 3.13 ng/mL; 1.57 ng/mL; 0 ng/mL).
- Pipette 150 µL of each sample per well in triplicate on a 96-well plate.
- Measure the fluorescence at an excitation peak of 428 nm and emission peak of 536 nm on a plate reader capable of reading fluorescence (see Table of Materials). Using statistical software (see Table of Materials), calculate the concentrations by interpolating the standard curve and using a four-parameter curve.
Representative Results
AO conformation
Enteroids suspended in 50% LWRN media for 72 h assume an AO conformation (Figure 1). This was confirmed via immunofluorescent staining utilizing enteroid whole mounts of the apical protein, zonula occludens-1 (ZO-1), and basolateral protein, β-catenin (Figure 1). AO enteroids show ZO-1 (green) on the outer, apical surface of the enteroid, while β-catenin (red) is on the inner, basolateral surface (Figure 1A). BO enteroids demonstrate the inverse with β-catenin (red) on the outer surface and ZO-1 (green) on the inner, luminal surface (Figure 1B). These results show the expected polarity reversal to confirm that the enteroids are AO prior to experimentation.
LY assay results
Increased permeability, demonstrated by the increased uptake of LY dye into the enteroid lumen, is expected in NEC6. BO enteroids treated with LPS and hypoxia showed significantly increased permeability compared to untreated controls using the technique described in this protocol (Figure 2A, **p = 0.005). This agrees with the literature that has described increased permeability in BO enteroid models of NEC4,13. Similarly, AO enteroids treated with LPS and hypoxia also demonstrated significantly increased permeability compared to untreated controls, as expected in an NEC model (Figure 3A, *p = 0.02). Treated AO enteroids showed increased uptake of LY in the lumen of treated enteroids (Figure 3C) compared to untreated controls (Figure 3B). This is similar to what is seen in BO enteroids, where LY is visualized in the enteroid lumen (Figure 2B). Increased LY dye uptake in the lumen of the enteroid results in increased fluorescence in treated enteroids, allowing for quantitative analysis via a microplate reader utilizing AO enteroids from different wells (Figure 3A). This demonstrates that this technique, utilizing LY, can be used to assess permeability in 3D enteroids. Outcomes can be analyzed using statistical software capable of interpolating a standard curve to determine the LY concentrations of the treatment groups. A Student's t-test, as shown in Figure 2 and Figure 3, can be used to determine the significance between groups.
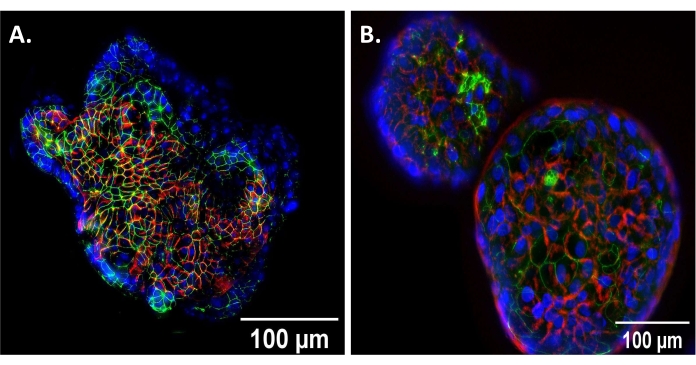
Figure 1: Verification of AO conformation. (A) Apical-out (AO) enteroid showing reverse polarity compared to (B) basolateral out (BO) enteroid showing traditional polarity at 20x magnification, scale bar set to 100 µm. DAPI, blue; beta-catenin (basolateral protein), red; zonula occludens-1 (apical protein), green. Please click here to view a larger version of this figure.
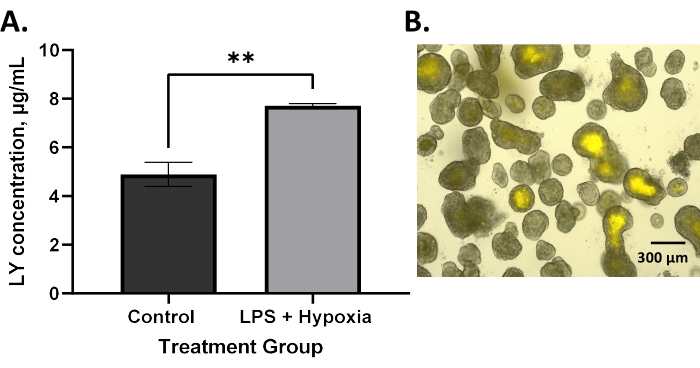
Figure 2: Increased permeability in a BO enteroid model after in vitro NEC induction as measured by an LY assay. (A) LPS and hypoxia-treated basolateral-out enteroids have significantly increased permeability compared to untreated controls using lucifer yellow assay; error bars show the standard error of the mean; **p = 0.005. (B) Image of basolateral-out enteroids with lucifer yellow visualized in the lumen following treatment of the media with lucifer yellow dye at 10x magnification, 300 µm scale bar. Please click here to view a larger version of this figure.
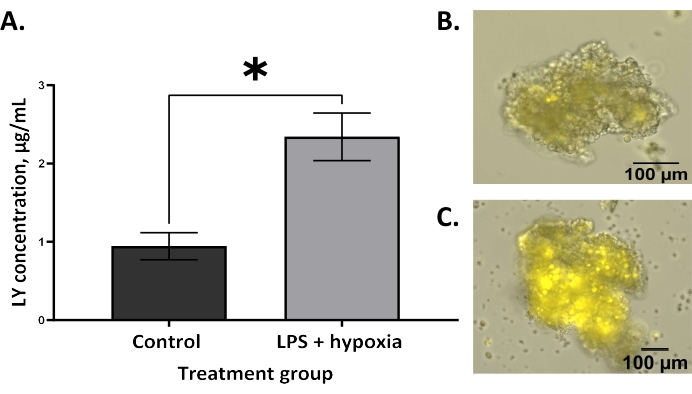
Figure 3: Increased permeability in an AO enteroid model after in vitro NEC induction as measured by an LY assay. (A) LPS and hypoxia-treated apical-out enteroids have significantly increased permeability compared to untreated controls using the lucifer yellow assay; error bars show the standard error of the mean; *p = 0.02. (B) A representative image of untreated control apical-out (AO) enteroid at 10x; the scale bar is set to 100 µm. (C) A representative image of an LPS and hypoxia-treated AO enteroid with LY is visualized in the lumen at 10x; the scale bar is set to 100 µm. Please click here to view a larger version of this figure.
Table 1: Composition of solutions, buffers, and media used in the present study. Please click here to download this Table.
Discussion
Intestinal permeability is complex and reflective of epithelial barrier function. The intestinal barrier comprises a single layer of epithelial cells that mediates transcellular and paracellular transport14. Paracellular permeability relies on tight junction proteins that seal the space between epithelial cells14. Within this paracellular transport, there are three distinct pathways by which molecules can cross: pore, leak, and unrestricted15. The pore pathway allows for permeability to small charged ions, while the leak pathway allows larger, uncharged molecules across15. The third, unrestricted pathway reflects permeability secondary to epithelial damage or death16. Although many molecules are used to assess paracellular permeability, the type and size of the molecule influence what permeability pathway will be investigated.
As a small, hydrophilic molecule, LY can cross through all three paracellular pathways, making it an optimal tool for studying paracellular permeability. In contrast, 4 kDa FITC-dextran, a sugar molecule used as a marker of paracellular permeability, can only traverse the leak and unrestricted pathways. The larger, 70 kDa FITC-dextran is limited to just the unrestricted pathway. Another method of determining paracellular permeability is via transepithelial resistance measurements (TEER), which measure electrical resistance across monolayers. This method only measures the pore pathway17. Thus, LY provides greater insight into the overall paracellular permeability by targeting all three pathways. This protocol capitalizes on this by using LY to study paracellular permeability.
The enteroid model was selected in this protocol as it more closely mimics in vivo conditions by creating a 3D mini-intestine to study. However, the challenge with the traditional BO enteroid model is how to access the apical surface to study host-pathogen interactions and the subsequent permeability changes. The AO enteroid model overcomes this challenge by reversing the polarity where the apical surface is in contact with the surrounding media. Several alternative methods of accessing the apical surface of enteroids have been described. These include gut-chip systems, microinjection, fragmentation, and growing enteroids in monolayers18,19. Advances in gut-chip systems have allowed the culturing of organoids and endothelial cells together in the context of vascularity and peristalsis to mimic the intestinal environment18. Microinjection involves the injection into the BO enteroid lumen via the use of specialized equipment such as a micromanipulator and microinjector19. Fragmentation uses the dissociation of enteroids into single cells that are then incubated in media with the pathogen before reforming a 3D structure19. The transformation of enteroids into 2D monolayers with subsequent treatment of the apical surface has also been described19. The AO enteroid model addresses some of the difficulties of these models by providing a simple, effective way to expose the apical surface without requiring expensive equipment or compromising the structural integrity of the enteroid.
Although using LY in an AO model allows for a broad study of paracellular permeability secondary to host-pathogen interactions, this protocol can be adapted to use with BO enteroids as well as FITC-dextran instead of LY. Two key changes to this protocol are required to modify it to be used for BO enteroids. First, BO enteroids can be maintained in BMM domes for all the wash steps before and after adding LY. It is important to use solutions warmed to 37 °C for these wash steps to prevent solubilization of the BMM dome. Second, the BMM dome needs to be disrupted with cold DPBS and scraping the well with a pipette tip after all the wash steps are complete in order to allow complete resuspension of the dissociated enteroids and LY. If FITC-dextran is substituted for LY, it is recommended to use the 4 kDa molecule as it crosses through two of the three paracellular pathways.
The key steps in this protocol include ensuring adequate washes to remove any trace LY from the solution outside the enteroid. It is also important to gently wash the enteroids to avoid dissociation and potential release of LY from the lumen into the surrounding solution until ready to quantitate using a microplate reader. Using warmed solutions during the wash steps also decreases stress on the enteroids. If cold solutions are used, enteroids may undergo apoptosis, leading to inaccurate results.
The limitations of this method include the inability to propagate AO enteroids and longer culturing times because of the additional 72 h required to reverse polarity. There is also a 2%-5% decrease in the number of treated AO enteroid compared to controls. Although this is overall negligible, it may lead to an underestimation of results. The limitations of using LY are similar to other permeability molecules, such as 4 kDa FITC-dextran, where trace amounts can cross the intestinal epithelial barrier via transcytosis20. Although this amount is negligible, it may affect the results. Based on these findings, applying LY to the media of AO enteroids provides an elegant and relatively simple way to quantitatively and qualitatively analyze paracellular bowel permeability in a 3D model.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
We would like to thank Ashley Nelson from the University of Rochester Medical Center for her instrumental help with our enteroid model. We would also like to thank the Division of Pediatric Surgery at the University of Oklahoma for their support of this project. This work was supported by the National Institute of Health [NIH Grant R03 DK117216-01A1], the Oklahoma Center for Adult Stem Cell Research, and the Presbyterian Health Foundation Grant #20180587 awarded to the Department of Surgery at the University of Oklahoma Health Sciences Center.
Materials
| [leu] 15-gastrin 1 | Millipore Sigma | G9145-.1MG | |
| 100 µm sterile cell strainer | Corning | 431752 | |
| 100% LWRN conditioned media | Made in-house following Miyoshi et al.12 | ||
| 24-well tissue culture plate | Corning | 3526 | |
| 96-well black, clear bottom plate | Greiner Bio-One | 655090 | |
| A-83-01 | R&D Systems | 2939/10 | |
| Alexa Fluor 488 goat anti-rabbit secondary ab, 1:1000 | Invitrogen | A-11034 | |
| Alexa Fluor 594 goat anti-mouse secondary ab, 1:1000 | Invitrogen | A-11032 | |
| Amphotericin B | Thermo Fisher Scientific | 15290026 | |
| Anti-zonula occludens-1 rabbit primary ab, 1:200 | Cell Signaling | #D6L1E | |
| Anti-β-catenin mouse primary ab, 1:100 | Cell Signaling | #14-2567-82 | |
| B-27 supplement minus Vitamin A | Thermo Fisher Scientific | 17504-044 | |
| Barrier PAP pen | Scientific Device Laboratory | 9804-02 | |
| BMM (Matrigel) | Corning | CB-40230C | |
| Cell Recovery Solution | Corning | 354270 | |
| Dissecting scissors | |||
| DMEM | Thermo Fisher Scientific | 11-965-118 | |
| DMEM/F-12 | Thermo Fisher Scientific | 11320-082 | |
| DPBS | Thermo Fisher Scientific | 14-190-144 | |
| Epidermal Growth Factor (EGF) | Millipore Sigma | GF144 | |
| Ethylenediaminetetraacetic acid (EDTA) | Millipore Sigma | EDS-500G | |
| EVOS m7000 Imaging system | Invitrogen | AMF7000 | |
| Fetal Bovine Serum (FBS) | Gemini Bio-Products | 100-525 | |
| Fluoroshield with DAPI | Millipore Sigma | F6057-20mL | |
| Forceps | |||
| Gentamicin | Thermo Fisher Scientific | 15-750-060 | |
| Glass coverslips | |||
| GlutaMAX | Thermo Fisher Scientific | 35050-061 | |
| GraphPad Prism 9 | Dotmatics | ||
| Insulin | Thermo Fisher Scientific | 12585014 | |
| Lipopolysaccharide (LPS) | Millipore Sigma | L2630-25MG | |
| Lucifer Yellow CH, Lithium Salt | Invitrogen | L453 | |
| Modular incubator chamber | Billups Rothenberg Inc. | MIC101 | |
| N-2 supplement | Thermo Fisher Scientific | 17502-048 | |
| N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) | Thermo Fisher Scientific | 15630-080 | |
| N-Acetylcysteine | Millipore Sigma | A9165-5G | |
| Nicotinamide | Millipore Sigma | N0636-100G | |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140-148 | |
| Refrigerated swinging bucket centrifuge | |||
| Refrigerated tabletop microcentrifuge | |||
| RPMI 1640 Medium | Thermo Fisher Scientific | 11875093 | |
| SB202190 | Millipore Sigma | S7067-5MG | |
| SpectraMax iD3 microplate reader | Molecular devices | ||
| Tube Revolver Rotator | ThermoFisher Scientific | 88881001 | |
| Ultra-low attachment 24-well tissue culture plate | Corning | 3473 | |
| Y-27632, ROCK inhibitor (RI) | Tocris | 1254 |
Referencias
- Ranganathan, S., Smith, E. M., Foulke-Abel, J. D., Barry, E. M. Research in a time of enteroids and organoids: How the human gut model has transformed the study of enteric bacterial pathogens. Gut Microbes. 12 (1), 1795492 (2020).
- De Fazio, L., et al. Necrotizing enterocolitis: Overview on in vitro models. International Journal of Molecular Sciences. 22 (13), 6761 (2021).
- Kovler, M. L., Sodhi, C. P., Hackam, D. J. Precision-based modeling approaches for necrotizing enterocolitis. Disease Models & Mechanisms. 13 (6), (2020).
- Ares, G. J., Buonpane, C., Yuan, C., Wood, D., Hunter, C. J. A novel human epithelial enteroid model of necrotizing enterocolitis. Journal of Visualized Experiments. (146), e59194 (2019).
- Co, J. Y., et al. Controlling epithelial polarity: A human enteroid model for host-pathogen interactions. Cell Reports. 26 (9), 2509-2520 (2019).
- Buonpane, C., et al. ROCK1 inhibitor stabilizes E-cadherin and improves barrier function in experimental necrotizing enterocolitis. The American Journal of Physiology-Gastrointestinal and Liver Physiology. 318 (4), 781-792 (2020).
- Hill, D. R., Huang, S., Tsai, Y. H., Spence, J. R., Young, V. B. Real-time measurement of epithelial barrier permeability in human intestinal organoids. Journal of Visualized Experiments. (130), e56960 (2017).
- Lian, P., Braber, S., Varasteh, S., Wichers, H. J., Folkerts, G. Hypoxia and heat stress affect epithelial integrity in a Caco-2/HT-29 co-culture. Scientific Reports. 11, 13186 (2021).
- Zhao, W., Han, L., Bae, Y., Manickam, D. S. Lucifer yellow – A robust paracellular permeability marker in a cell model of the human blood-brain barrier. Journal of Visualized Experiments. (150), e58900 (2019).
- Manabe, A., et al. Chlorpheniramine increases paracellular permeability to marker fluorescein lucifer yellow mediated by internalization of occludin in murine colonic epithelial cells. Biological and Pharmaceutical Bulletin. 40 (8), 1299-1305 (2017).
- Bardenbacher, M., et al. Permeability analyses and three dimensional imaging of interferon gamma-induced barrier disintegration in intestinal organoids. Stem Cell Research. 35, 101383 (2019).
- Miyoshi, H., Stappenbeck, T. S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nature Protocols. 8 (12), 2471-2482 (2013).
- Buonpane, C., et al. Experimental modeling of necrotizing enterocolitis in human infant intestinal enteroids. Journal of Investigative Surgery. 35 (1), 111-118 (2022).
- Chanez-Paredes, S. D., Abtahi, S., Kuo, W. -. T., Turner, J. R. Differentiating between tight junction-dependent and tight junction-independent intestinal barrier loss in vivo. Methods in Molecular Biology. 2367, 249-271 (2021).
- Shen, L., Weber, C. R., Raleigh, D. R., Yu, D., Turner, J. R. Tight junction pore and leak pathways: A dynamic duo. Annual Review of Physiology. 73, 283-309 (2011).
- Monaco, A., Ovryn, B., Axis, J., Amsler, K. The epithelial cell leak pathway. International Journal of Molecular Sciences. 22 (14), 7677 (2021).
- Srinivasan, B., et al. TEER measurement techniques for in vitro barrier model systems. Journal of Laboratory Automation. 20 (2), 107-126 (2015).
- Kasendra, M., et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Scientific Reports. 8, 2871 (2018).
- Stroulios, G., et al. Culture methods to study apical-specific interactions using intestinal organoid models. Journal of Visualized Experiments. (169), e62330 (2021).
- Frost, T. S., Jiang, L., Lynch, R. M., Zohar, Y. Permeability of epithelial/endothelial barriers in transwells and microfluidic bilayer devices. Micromachines. 10 (8), 533 (2019).
