Detection of Post-Replicative Gaps Accumulation and Repair in Human Cells Using the DNA Fiber Assay
Summary
Here we describe two modifications of the DNA fiber assay to investigate single-stranded DNA gaps in replicating DNA after lesion induction. The S1 fiber assay enables the detection of post-replicative gaps using the ssDNA-specific S1 endonuclease, while the gap-filling assay allows visualization and quantification of gap repair.
Abstract
The DNA fiber assay is a simple and robust method for the analysis of replication fork dynamics, based on the immunodetection of nucleotide analogs that are incorporated during DNA synthesis in human cells. However, this technique has a limited resolution of a few thousand kilobases. Consequently, post-replicative single-stranded DNA (ssDNA) gaps as small as a few hundred bases are not detectable by the standard assay. Here, we describe a modified version of the DNA fiber assay that utilizes the S1 nuclease, an enzyme that specifically cleaves ssDNA. In the presence of post-replicative ssDNA gaps, the S1 nuclease will target and cleave the gaps, generating shorter tracts that can be used as a read-out for ssDNA gaps on ongoing forks. These post-replicative ssDNA gaps are formed when damaged DNA is replicated discontinuously. They can be repaired via mechanisms uncoupled from genome replication, in a process known as gap-filling or post-replicative repair. Because gap-filling mechanisms involve DNA synthesis independent of the S phase, alterations in the DNA fiber labeling scheme can also be employed to monitor gap-filling events. Altogether, these modifications of the DNA fiber assay are powerful strategies to understand how post-replicative gaps are formed and filled in the genome of human cells.
Introduction
Seminal works have provided evidence of the accumulation of post-replicative single-stranded (ssDNA) gaps upon treatment with DNA damaging agents in bacteria1 and human cells2,3. During replication of damaged DNA templates, the DNA synthesis machinery may bypass the lesions by employing specific translesion synthesis DNA polymerases or through template switching mechanisms. Alternatively, the replisome may also simply skip the lesion leaving an ssDNA gap behind, to be repaired later. More recently, a study clearly showed that treatment with genotoxic agents leads to ssDNA gaps in eukaryotes by utilizing electron microscopy to visualize the specific structure of replication intermediates4. The formation of these regions of post-replicative gaps was initially proposed to be a simple result of the semi-discontinuous mode of DNA replication2. In this case, a lesion on the lagging strand can block the elongation of an Okazaki fragment, but fork progression is naturally rescued by the following Okazaki fragment, leaving behind an ssDNA gap. However, further studies demonstrated that the formation of gaps on the leading strand is also possible, as first shown in bacteria5. In eukaryotes, PRIMPOL, a unique DNA polymerase with primase activity, was shown to be able to restart DNA synthesis downstream a replication blocking lesion through its repriming activity6,7,8,9,10,11. Thus, the PRIMPOL primase activity may explain the formation of post-replicative ssDNA gaps in the leading strand upon treatment with a DNA damaging agent in human cells12. Nonetheless, detection of these gaps as well as gap repair, until recently, required indirect or time-consuming approaches such as electron microscopy4 or plasmid-based assays13. The use of the ssDNA-specific S1 nuclease to detect gaps in human cells was pioneered by early studies more than forty years ago, using sucrose gradient techniques2,3. More recently, our group applied the use of this nuclease to detect ssDNA in replicating DNA (post-replicative gaps) using other methods such as DNA fiber and comet assays14. These new approaches paved the way for the current surge of studies on post-replicative gaps. Here, we describe a strategy of using S1 nuclease to detect post-replicative ssDNA gaps by DNA fiber assay and explain how a differential labeling scheme in the DNA fiber protocol can permit the study of the repair of these gaps.
The DNA fiber assay is a powerful technique that has been used by a growing number of labs and has provided valuable insights into replication fork dynamics and replication stress response mechanisms. Briefly, this technique is based on the sequential incorporation of nucleotide analogs (such as CldU -5-chloro-2'-deoxyuridine-, and IdU -5-iodo-2'-deoxyuridine) in the replicating DNA. After harvesting, cells are lysed, and DNA molecules spread on a positively coated glass slide. CldU and IdU are then detected by specific antibodies, which can be visualized in a fluorescent microscope as bicolored fibers. Finally, the lengths of the IdU and CldU tracts are measured to identify any alterations of DNA replication dynamics as a consequence of DNA damage induction. This technique can be utilized to investigate different phenomena, such as fork stalling, fork slowing, nascent DNA degradation, and variations in the frequency of origin firing15,16.
One limitation of the DNA fiber assay is its resolution of a few kilobases. Because post-replicative ssDNA gaps can be in the range of hundreds of bases, it is impossible to visualize these gaps directly by the standard DNA fiber protocol. The presence of ssDNA gaps on replicating DNA in human cells treated with genotoxic agents has been indirectly implicated before. For example, the observation of ssDNA as assessed by recruitment of the ssDNA-binding protein, replication protein A (RPA), in cells outside the S phase, or the formation of ssDNA as detected by alkaline DNA unwinding combined with the absence of prolonged fork stalling by DNA fiber assay12,17,18,19,20,21 was attributed to the accumulation of ssDNA gaps. In addition, post-replicative ssDNA gaps induce an ATR-dependent G2/M phase checkpoint and arrest cells treated with replication poisons18,19,20,21,22.
The S1 nuclease degrades single-stranded nucleic acids releasing 5'-phosphoryl mono- or oligonucleotides and has a 5-times higher affinity to ssDNA compared to RNA. Double-stranded nucleic acids (DNA:DNA, DNA:RNA, or RNA:RNA) are resistant to the S1 nuclease except when used in extremely high concentrations. The S1 nuclease also cleaves double-stranded DNA at the single-stranded region caused by a nick, gap, mismatch, or loop. The S1 nuclease is thus an ssDNA-specific endonuclease capable of cleaving ssDNA gaps, ultimately generating double-stranded breaks23,24. Thus, adding steps for S1 nuclease digestion to the DNA fiber protocol indirectly enables the detection of post-replicative ssDNA gaps. In the presence of gaps, treatment of exposed nuclei with the S1 nuclease prior to DNA spreading will generate shorter tracts as a consequence of S1 cleavage of ssDNA inherently present at gaps14. Accordingly, tract shortening is the read-out for ssDNA gaps using this approach. Compared to the standard DNA fiber protocol, the DNA fiber with the S1 nuclease only requires two extra steps: nuclei exposure (cell permeabilization) and treatment with the S1 nuclease. It is important to note that appropriate controls are mandatory, such as samples treated with the genotoxic agent but without the S1 nuclease and samples treated with the S1 nuclease without the genotoxic agent. The protocol itself, including the incorporation of analogs, S1 treatment, and spreading, can be performed in one day and does not require exceptional material. It only requires the thymidine analogs, the purified S1 nuclease, the appropriate primary and secondary antibodies, and a fluorescent microscope. Overall, the DNA fiber employing the S1 nuclease detects ssDNA gaps on ongoing replication forks using a relatively simple approach.
The post-replicative ssDNA gaps formed as a consequence of replication stress response mechanisms can be repaired (or filled) by different mechanisms, including translesion DNA synthesis or template switching, in a process called gap-filling or post-replication repair (PRR)25. These processes occur behind the advancing forks, involving replication-independent DNA synthesis14,26,27. Based on these findings, a labeling scheme distinct from the standard DNA fiber assay can be performed to visualize gap-filling events in the G2 phase directly14,16,26,28. Specifically, one thymidine analog can be used to label the replication fork at the time of genotoxic treatment and post-replicative ssDNA gap formation, while another thymidine analog can be used to label gap-filling events. In this protocol, cells are labeled with a first thymidine analog (IdU, for example) immediately after or concomitantly with genotoxic treatment for 1 h, so that the nascent DNA is labeled at the time of gap formation. Nocodazole is added upon treatment for anywhere between 12-24 h to arrest cells in G2/M, preventing the following S phase. For the last 4 h of nocodazole treatment, a second thymidine analog (CldU, for example) is added to the media to be incorporated during gap filling. Importantly, this assay can only be used to detect gap-filling events in G2 because the gap-filling signal from CldU cannot be distinguished from a signal due to CldU incorporation into replicating DNA in the S phase. Therefore, to minimize the background signal, the timing of CldU incorporation after damage induction should coincide with when most of the cell population is entering the G2 phase14. Therefore, this timing will vary depending on cell line and treatment conditions. Optimizing cell cycle progression prior to employing this assay is advised. Co-staining of these thymidine analogs allows the visualization of gap-filling (PRR) tracts (CldU patches) on top of nascent DNA (IdU tracts) synthesized during the genotoxic treatment when ssDNA gaps were generated.
Protocol
As the study uses human cells, the work was approved by the Ethics Committee of the Institute of Biomedical Science at the University of São Paulo (ICB-USP, approval number #48347515.3.00005467) for the research with human samples.
NOTE: The protocols described here were used in previous publications with minor modifications14,16,28. Here the focus is on the use of ultraviolet light C (UVC) as a DNA damaging agent. However, other DNA damaging agents such as cisplatin and hydroxyurea have also been successfully employed28. This protocol can be performed in an array of cell lines, including U2OS, HEK293T, human fibroblasts, and others14,28,29. It is essential to determine the experimental question before performing this protocol. The DNA fiber assay with the S1 nuclease (called S1 Fiber hereafter) is used to detect the presence of post-replicative ssDNA gaps, while the gap-filling (or PRR) assay is performed to quantify gap-filling events in the G2 phase. Thus, if analyzing the presence of post-replicative ssDNA gaps, the investigator should perform section 2 (S1 Fiber experimental setup) and proceed directly to section 4 (DNA fiber preparation by spreading) of the protocol. If analyzing gap-filling events, the investigator should proceed directly to section 3 (Gap-filling (PRR) experimental setup).
1. Reagents and setup
- Thymidine analogs: Resuspend 5-Iodo-2'-deoxyuridine (IdU) and 5-Chloro-2'-deoxyuridine (CldU) in 1 N NH4OH to a final concentration of 100 mM. Filter the analogs through a sterile syringe filter (0.2 µm), aliquot, and store them at -20 C.
- Antibodies: Primaries – anti-BrdU rat (Biorad) and anti-BrdU mouse (BD); Secondaries – anti-rat Alexa Fluor 488 and anti-mouse Alexa Fluor 594.
- Cellular permeabilization CSK100 buffer for the S1 Fiber: Add 100 mM NaCl, 10 mM MOPS [pH 7], 3 mM MgCl2 [pH 7.2], 300 mM sucrose and 0.5% Triton X-100 in distilled H2O.
- S1 nuclease buffer [pH 4.6]: Add 30 mM Sodium Acetate [pH 4.6], 10 mM Zinc Acetate, 50 mM NaCl and 5% Glycerol in H2O.
NOTE: The S1 nuclease buffer must have a final pH of 4.6, as the S1 activity drops 50% at pH > 4.9. - Aliquot and store at -20 °C the S1 nuclease purified from A. oryzae pre-diluted in S1 nuclease dilution buffer provided by the manufacturer.
- Reconstitute the nocodazole in DMSO to a final concentration of 2 mM for gap-filling. assay.
- Lysis buffer: Add 200 mM Tris-HCl [pH 7.5], 50 mM EDTA and 0.5 % SDS in H2O.
- Prepare phosphate-buffered saline (PBS).
- Prepare PBS-T by adding 0.1% Tween-20 in PBS.
- Prepare 5% Bovine Serum Albumin (BSA) in PBS.
- Prepare 0.1% BSA in PBS.
- Prepare PBS-T-BSA by adding 1% BSA in PBS-T.
NOTE: Use an epifluorescent microscope with a mercury or xenon lamp with a 510 nm wavelength and 42 nm bandwidth GFP (FITC, Alexa Fluor 488) emission filter and a 624 nm wavelength and 40 nm bandwidth Texas Red (Alexa Fluor 594) emission filter or a confocal microscope with a 488-nm line (blue) laser to detect green fluorescence (detected between 510 and 560 nm) and a 561-nm line (yellow) laser to detect red fluorescence (detected between 575 and 680 nm). Immersion objective (40x, 63x or 100x).
2. S1 Fiber experimental setup (2 days)
- Day 1: Plate 4 wells for each cell line: ± UVC and ± S1 nuclease with cells at 70-90% confluency.
NOTE: The experiment can be done in a format as small as a 12-well plate. In this case, all solutions are used at 0.5 mL/well. - Day 2: Prepare fresh IdU at 20 µM and CldU at 200 µM in pre-warmed (37 °C) cell culture media.
- Aspirate the culture media and immediately add the media with 20 µM IdU and incubate the cells at 37 °C, 5% CO2 (cell incubator) for precisely 20 min.
- Quickly wash the cells with pre-warmed (37 °C) PBS twice.
- Irradiate the cells with 20 J/m2 UVC. Use untreated cells as controls.
- Immediately add the media with 200 µM CldU and incubate the cells at 37 °C, 5% CO2 (cell incubator) for precisely 60 min.
NOTE: IdU and CldU labeling can be interchanged, but the concentration of the second analog should be 5-10 times higher than the first analog. Timing of the analog incubation can be adapted to the genotoxic agent used, but the time of the second analog incubation should be long enough to ensure enough time for gap formation14,16. If using a DNA damaging drug, add the drug together with CldU28,29,30. - Wash the cells with pre-warmed (37 °C) PBS twice.
- Permeabilize the cells with the CSK100 buffer for 8-10 min at room temperature (RT).
NOTE: Successful permeabilization can be checked under a brightfield microscope where only nuclei should be observable - Carefully wash the nuclei with PBS (pour 0.5 mL down the side of each well, just for a few seconds).
- Carefully wash the nuclei with S1 buffer (pour 0.5 mL down the side of each well, just for a few seconds).
- Incubate the nuclei with S1 buffer with S1 nuclease (20 U/mL) or without (as a control) for 30 min at 37 °C (cell incubator).
- Aspirate the S1 buffer and add 0.1% BSA in PBS.
- Scrape the nuclei and transfer them to an appropriately annotated 1.5 mL tube. Keep tubes on ice all the time.
NOTE: Successful detachment of nuclei can be checked under a brightfield microscope. - Pellet the nuclei: centrifuge at ~4600 x g for 5 min, at 4 °C.
NOTE: Different from the standard DNA fiber assay, the nuclei cannot be counted, so consider the initial number of cells plated. Alternatively, an extra well that was not subjected to permeabilization can be used to estimate the cell count using a hemocytometer or a cell counter. - Resuspend the pellets well in PBS at a concentration of ~1500 cells/µL, place the tubes on ice and proceed to DNA fiber preparation by spreading (section 4) protocol immediately
3. Gap-filling (PRR) experimental setup (3 days)
- Day 1: Plate 2 wells for each cell line: ± UVC with cells at ~70 % confluency.
- Day 2: Prepare fresh IdU at 20 µM in pre-warmed (37 °C) cell culture media.
- Irradiate the cells with 20 J/m2 UVC.
- Immediately add the media with 20 µM IdU and incubate the cells at 37 °C, 5% CO2 (cell incubator) for precisely 60 min.
- Wash the cells with pre-warmed (37 °C) PBS twice.
- Immediately add the media with 200 ng/mL nocodazole and incubate the cells at 37 °C, 5% CO2 (cell incubator) for 12-24 h (timing of this step must be kept consistent between experiments).
NOTE: The concentration of nocodazole might need to be adapted according to the cell line used. If using a genotoxic drug, incubate the cells with IdU ± drug28. - Day 3: Aspirate the culture media, add the media with 20 µM CldU + nocodazole, and incubate for the last 4 h of nocodazole treatment, at 37 °C, 5% CO2 (cell incubator).
- Wash the cells with pre-warmed (37 °C) PBS.
- Add trypsin and incubate for 1-2 min at 37 °C, 5% CO2 (cell incubator) to detach cells.
- Add the same volume of cell culture media and collect cells (media + trypsin) in an appropriately annotated 1.5 mL tube. Keep the tubes on ice all the time.
- Count the cells using a hemocytometer or a cell counter.
- Pellet the cells: centrifuge at ~350 x g for 5 min, at 4 °C.
- Remove the supernatant and resuspend the cells in PBS at ~1500 cells/µL. Keep the cells on ice and start DNA fiber preparation by spreading protocol immediately (Day 3 or 4).
4. DNA fiber preparation by spreading (~1 h for 12 slides)
- Annotate the microscope slides with a carbon pencil and place them flat on a tray.
- Tap the bottom of the tube to ensure homogenization.
- Add 2 µL of cells on the top of the corresponding slide.
NOTE: Two drops can be added, one on the top of the slide and another in the middle of the slide. - Add 6 µL of the lysis buffer and lyse the cells by pipetting up and down about 5 times (avoid making bubbles).
- Pull the drop a little bit towards the bottom of the slide with the pipette tip (to guide the path of the drop spreading).
- Incubate for 5 min at RT to lyse the nuclei.
NOTE: The volume of lysis buffer and the time of lysis can be adjusted if the lysate dries too quickly or, alternatively, does not dry enough. Lysis buffer volume can vary from 5-7 µL and timing can fall between 4-10 min. - Tilt the slide at around 20-40° and allow the drop to spread to the bottom of the slide at a constant, low speed.
- Allow the slide to dry at RT in the dark (10-15 min).
- Freshly prepare the fixing solution: Mix methanol: glacial acetic acid at a 3:1 ratio.
- Immerse the slides in a jar containing the fixing solution and incubate for 5 min at RT to fix DNA onto the slides.
CAUTION: Methanol is highly toxic and should be kept under a chemical hood. Discard in an appropriate container. - Dry the slides at RT in the dark and store at 4 °C protected from light until staining.
5. Immunostaining of DNA fibers (~6 h)
- Wash the slides with PBS twice for 5 min.
- Denature the DNA with freshly prepared 2.5 M HCl for 40-60 min at RT.
CAUTION: To prepare 2.5 M HCl, pour acid in water and never the opposite. This is an exothermic reaction so it will warm up slightly. - Wash the slides with PBS three times for 5 min.
- Block with 5% BSA (can be stored at 20 °C for up to three uses) previously warmed at 37 °C for 30-60 min at 37 °C.
- Remove the excess liquid from slides by gently tapping on a paper.
- Add 30 µL of primary antibodies (mouse anti-BrdU 1/20 (for IdU) and rat anti-BrdU 1/100 (for CldU) diluted in PBS-T-BSA) to the slides dropwise and place a coverslip on top. Incubate for 1-1.5 h at RT in a dark, humid chamber.
- Place the slides in a jar with PBS for 1-2 min, then carefully remove the coverslips.
- Wash with PBS-T in a jar three times for 5 min, then place the slides in PBS.
- Remove the excess liquid by gently tapping on a paper.
- Add 30 µL of secondary antibodies (anti-mouse Alexa Fluor 594 and anti-rat Alexa Fluor 488 diluted at 1/75 in PBS-T-BSA.) to the slides dropwise and place a coverslip on top. Incubate for 45-60 min at RT in a dark, humid chamber.
- Place the slides in a jar with PBS for 1-2 min, then carefully remove the coverslips.
- Wash with PBS-T in a jar three times for 5 min, then place the slides in PBS.
- Remove the excess liquid by gently tapping on a paper.
- Add 20 µL of the mounting reagent dropwise to the slides and place a coverslip on top. Avoid bubbles.
- Let the slides dry at RT in the dark, then store at 4 °C (or -20 °C for longer conservation).
6. Image acquisition and analysis (~1 h per sample)
NOTE: An epifluorescence microscope can be used for both protocols. However, for the gap-filling (PRR) assay, using a confocal microscope is highly recommended for the increased resolution of the images.
- Place a drop of immersion oil (microscope specific) near the top of the slide where the cells were lysed and find the focus using the green channel by searching green dots in the background (40x, 63x, and 100x objectives can be used).
- Find the main concentration of fibers and move to the edges to find well-spread non-overlapping individual fibers using only one channel to select the regions for the pictures and avoid potential bias.
- Take around 15-20 pictures per slide alongside the entire slide at the green and red channels and merge the two channels to obtain bicolor DNA fibers.
NOTE: If using a confocal microscope, take pictures at high resolution (1024 x 1024 pixels) and use pinhole values close to 1 for both red and green fluorescence. - Use ImageJ (free software provided by the NIH, https://imagej.nih.gov/ij/) for IdU and CldU tract length measurements.
- Open a picture by dragging it into the ImageJ box.
- Use the scale bar generated by the microscope software (appropriately calibrated) to convert the length to micrometers. Use the Straight-line tool by selecting the 5th icon from left to right on the ImageJ box to measure the scale bar in the picture provided micrometers and obtain a measurement in pixels. Click on Analyze > Set Scale and replace the "distance in pixels" with the appropriate number and the "unit of length" to µm.
- On the ImageJ box, select the Segmented Line tool by left-clicking on the 5th icon from left to right and then selecting the second option from top to bottom.
- Draw the exact path of the red (IdU) or green (CldU) tract by left-clicking and stop the drawing by right-clicking. Push the M button on the keyboard to measure the length.
- For the S1 Fiber, only analyze red-green (bicolor fibers)16. Measure red and green tract lengths from at least 150 fibers from different pictures by keeping track of the red and green tracts measurements for each individual fiber.
- For the gap-filling assay, only measure the length of IdU (red) tracts that contain at least 1 CldU (green) patch but no continuous CldU staining. Score at least 10-15 IdU tracts from different pictures with at least 1 CldU patch.
NOTE: Other software can be used for the analyses, such as Zeiss LSM Image Browser.
- For the S1 Fibers, plot the IdU tracts length, CldU tracts length, and ratios (CldU/IdU or vice-versa) in separate graphics as scatter dot plots with medians.
- Measure gap-filling events as units of "per-kilobase density", divide the total number of CldU (PRR, green) patches on a given length of IdU (red) tract by the total length of the red tract in kilobases (considering 1 µm = 2.59 kilobases31). Then plot the data as scatter dot plots with medians.
- In both experiments, perform statistical analysis between two samples using Mann-Whitney (nonparametric test) and between more than two samples using Kruskal-Wallis followed by Dunn's multiple comparisons test.
Representative Results
In the S1 Fiber assay, if treatment with a genotoxic agent leads to post-replicative ssDNA gaps, the overall lengths of DNA fibers from S1-treated nuclei will be shorter upon treatment with DNA damage compared to untreated samples as well as to samples that were treated with the genotoxic agent but were not submitted to S1 cleavage (Figure 1).
Alternatively, if treatment with the S1 nuclease does not significantly affect the length of DNA fibers compared to untreated cells, different interpretations are possible: (1) There is no formation of post-replicative ssDNA gaps upon treatment with the genotoxic agent used, and/or in the specific genetic background investigated. Indeed, we have previously shown that UV-induced 6-4 photoproducts (6-4PP) but not cyclobutane pyrimidine dimers (CPD) lead to the formation of ssDNA gaps in cells deficient for nucleotide excision repair (NER, XP-C cells)14. (2) The ssDNA gaps were repaired and can no longer be detected. (3) The shortening induced by treatment with the genotoxic agent alone is too pronounced to allow detection of a further shortening via S1 cleavage.
Notably, the lack of effect of the S1 might also reflect technical issues such as impaired conditions for the S1 activity, including inappropriate pH of the S1 buffer or limited amount of zinc.
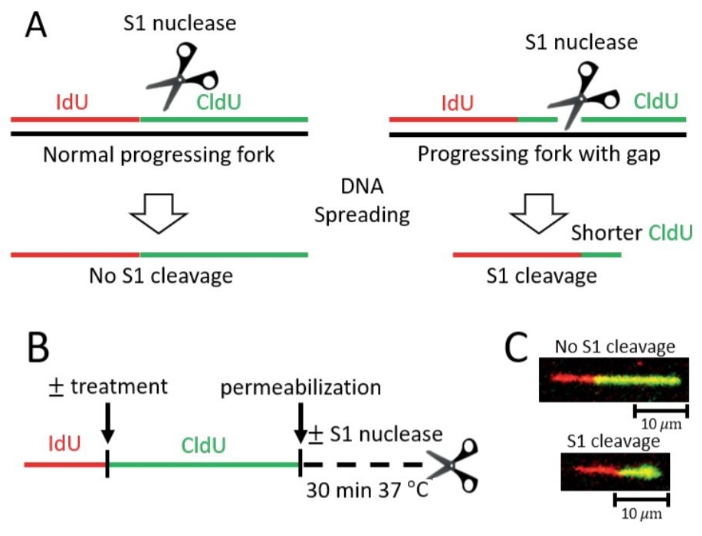
Figure 1: Detection of post-replicative ssDNA gaps on ongoing forks by S1 Fiber assay. (A) Schematic representation of DNA fiber assays with the ssDNA specific S1 nuclease (S1 Fiber). Progressing replication forks are labeled with thymidine analogs (IdU, CldU) and can be measured following DNA spreading and immunostaining. Left, standard progressing forks without gaps are not susceptible to cleavage by the S1 nuclease. Right, gaps in nascent DNA that are conventionally undetectable will be targeted by S1 nuclease and will be visualized as shorter CldU tracts. (B) Scheme of the S1 Fiber protocol. (C) Representative images of S1 Fiber. Top, control nascent DNA tract without gaps, impervious to cleavage by the S1 nuclease. Bottom, DNA tract positive for gaps cleaved by the S1 nuclease resulting in a shorter CldU (green) tract length. Please click here to view a larger version of this figure.
In the gap-filling (PRR) assay, a significant increase in the density of short CldU patches on IdU tracts is indicative of increased gap-filling events (Figure 2). A high density of PRR events can be reflected by 1 PRR event (green/CldU patch) in a short IdU tract as well as by multiple PRR events in a long tract (see representative images Figure 2C). The analysis must be rigorously performed by scoring only CldU patches that are on top of IdU tracts considering that the CldU patches are small and could easily be mistaken as staining background (see representative images Figure 2C).
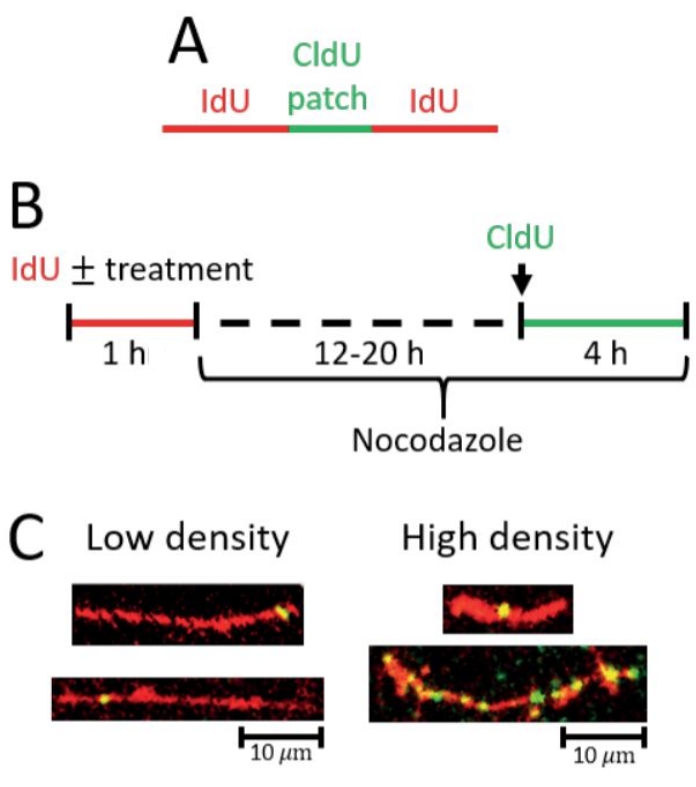
Figure 2: Detection of post-replicative repair by gap filling. (A) Scheme of red (IdU) tract with green (CldU) patch indicative of a gap-filling event. (B) Scheme of the protocol used to detect gap-filling. (C) Representative images from gap-filling (PRR) assay. Left, longer IdU tracts with at least one CldU patch and/or fewer CldU patches total correspond with low gap-filling density (fewer gap-filling events). Right, shorter IdU tracts with at least one CldU patch and longer IdU tracts with more CldU patches total correspond with high gap-filling density (more gap-filling events). Please click here to view a larger version of this figure.
Discussion
Critical steps of the standard DNA fiber assay protocol were discussed in a previous publication32. Here, we describe modified versions of the standard DNA fiber assay to investigate the presence of post-replicative ssDNA gaps as well as their repair by gap-filling, initially described in14. In the context of post-replicative ssDNA gap presence, the use of the S1 nuclease in the S1 Fiber protocol would most likely be suitable after exposure of a genotoxic agent for a minimum of 1 h to allow time for gap formation and subsequent detection. However, it is essential to note that in some cell lines or genetic backgrounds, the use of the S1 nuclease might generate shorter tracts even without additional treatment with genotoxic agents33,34. Therefore, it is crucial to include all the controls to ensure proper conclusion on whether treatment with a genotoxic agent leads to gap accumulation. In addition, it is important to further validate the S1 Fiber result by repeating the assay in conditions in which ssDNA gaps are no longer formed or are repaired.
Regarding the gap-filling (PRR) assay, it is important to optimize a UVC dose/drug concentration that does not affect the overall length of the DNA fibers, as results are calculated as a per-kilobase density. If using a concentration that induces tract shortening, results may be artificially skewed towards an increased gap-filling density, making data interpretation difficult. To counteract this limitation, gap-filling events can be visualized on top of non-labeled DNA by staining total DNA using an anti-ssDNA antibody, as previously described14,16. Furthermore, a kinetic experiment might be performed to ensure maximum detection of gap-filling events.
Note that gap-filling events can also be monitored using the S1 Fiber assay when cells are allowed to recover before treatment with the S1 nuclease. Specifically, when post-replicative gaps are filled, DNA fibers become insensitive to cleavage by the S1 nuclease, and DNA fiber length can therefore be used as a read-out for gap filling in this context28. Thus, this approach can be used to study gap-filling throughout the cell cycle, differently from the PRR assay that is limited to the investigation of gap filling in G2/M.
Finally, data obtained with the PRR assay were validated using the S1 Fiber28, suggesting that nocodazole and artificial blockage of cells in G2/M during the PRR assay do not affect gap filling. Other agents that induce G2/M phase blockage can potentially be used in this assay
The S1 Fiber has been increasingly used by labs worldwide, providing new insights into the mechanisms of ssDNA gaps formation and revealing previously unappreciated conditions in which these gaps can be formed14,29,34,35,36. For future investigations, it would be interesting to combine these modified protocols with the quantum dot approach that allows direct visualization of the DNA lesion37 to elucidate the cross-talk between post-replicative ssDNA gap formation and DNA repair as well as how the nature of the damage defines the mechanism of the replication stress response.As an alternative to the S1 Fiber assay, the S1 nuclease can also be used to study the gap-filling process in a modified neutral comet assay. Briefly, if gaps are present, treatment of comets with the S1 nuclease leads to the formation of double-stranded breaks, detectable by neutral comet assay. Protocols for neutral comet assay as well as enzyme-modified comet assay were previously published38,39. In particular, this approach allows for ssDNA gap detection in all phases of the cell cycle, with no need to arrest cells in G2 phase. Using this approach, combined with the S1 Fiber and gap-filling (PRR) protocols described here, we found that REV3L (DNA polymerase zeta catalytic unit) is responsible for filling gaps formed when UV-induced 6-4PPs are present in the human genome14.
With the limited available technical approach, post-replicative ssDNA gap formation and, in particular, gap-filling events have remained largely unexplored in human cells. Altogether, these modified techniques (S1 Fiber, gap-filling (PRR) assay, and S1 Comet) provide novel strategies for investigating the presence of ssDNA gaps and how these gaps are ultimately filled in the human genome.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
The work in C.F.M.M. laboratory is supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, São Paulo, Brazil, Grants #2019/19435-3, #2013/08028-1 and 2017/05680-0) under the International Collaboration Research from FAPESP and The Netherlands Organization for Scientific Research (NWO, The Netherlands); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, DF, Brazil, Grants # 308868/2018-8] and Coordenação de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES, Brasília, DF, Brazil, Finance Code 001).
Materials
| Acetic acid, Glacial | Synth | 64-19-7 | Alternatively, BSA – Biosera – REF PM-T1725/100 |
| Ammonium hydroxide | Synth | 1336-21-6 | Or similar |
| Antibody anti-mouse IgG1 Alexa Fluor 594 | Invitrogen | A11005 | – |
| Antibody anti-rat Alexa Fluor 488 | Invitrogen | A21470 | – |
| Antibody Mouse anti-BrdU | Becton Disckson | 347580 | – |
| Antibody Rat anti-BrdU | Abcam | Ab6326 | – |
| Biological security hood | Pachane | PA 410 | Use hood present in the laboratory |
| BSA (Bovine Serum Albumin) | Sigma-Aldrich | A3294 | Or similar |
| Cell scraper | Thermo Scientific | 179693 | Or similar |
| CldU | Millipore-Sigma | C6891 | – |
| Cloridric acid | Synth | 7647-01-0 | Or similar |
| Confocal Zeiss LSM Series (7, 8 or 9) | Zeiss | – | Or similar |
| Cover glass (or coverslips) | Thermo Scientific | 152460 | Alternatively, Olen – Kasvi Cover Glass (24 x 60 mm) – K5-2460 |
| DMEM – High Glucose | LGC/Gibco | BR30211-05/12100046 | Use culture media specific for the cell line used. |
| EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate) | Sigma-Aldrich | E5314 | Or similar |
| Epifluorescence Microscope Axiovert 200 | Zeiss | – | Or similar |
| FBS (Fetal Bovine Serum) | Gibco | 12657-029 | Or similar |
| Forma Series II Water Jacketed CO2 Incubator | Thermo Scientific 3110 | 13-998-074 | Use cell incubator present in the laboratory |
| Glass slide jar | Sigma-Aldrich | S5516 | Or similar |
| Glycerol | Sigma-Aldrich | 56-81-5 | Or similar |
| Idu | Millipore-Sigma | I7125 | – |
| Magnesium Chloride | Synth | 7791-18-6 | Or similar |
| Methanol | Merck | 67-56-1 | Or similar |
| Microscope slides | Denville | M1021 | Alternatively, Olen – Kasvi Microscope Slides – K5-7105 OR Precision Glass Line – 7105-1 |
| MOPS (Ácido 3-morfolinopropano 1-sulfônico) | Synth | 1132-61-2 | Or similar |
| Nocodazole | Sigma-Aldrich | 31430-18-9 | – |
| PBS (Phosphate Buffer Saline) | Life Thechnologies | 3002 | Or similar |
| Penicillin-Streptomycin | Gibco | 15140122 | Or similar |
| ProLong Gold AntiFade Mountant | Invitrogen | P36930 | Any antifade moutant solution for immunofluorescence could be used |
| S1 nuclease purified from Aspergillus oryzae | Invitrogen | 18001-016 | Pre-dilute the S1 nuclease (1/100 – 1/200) in S1 nuclease dilution buffer provided by the manufacturer, aliquote and store at -20 °C |
| SDS (Sodium Dodecyl Sulfate) | BioRad | 161-0302 | Or similar |
| Sodium Acetate Trihydrate | Sigma-Aldrich | 6131-90-4 | Or similar |
| Sodium Chloride | Synth | 7647-14-5 | Or similar |
| Sucrose | Sigma-Aldrich | 57-50-1 | Or similar |
| Tris Base | West Lab Research | BP152-1 | Or similar |
| Triton X-100 | Synth | 9002-93-1 | Or similar |
| Trypsin | Gibco | 25200072 | Or similar |
| Tween 20 | Sigma-Aldrich | P1379 | Or similar |
| UVC Lamp | Non Specific | – | Essential: emission lenght of 254 nm |
| VLX-3W UV Radiometer | Vilber Loumart | – | Or similar |
| Zinc Acetate | Sigma-Aldrich | 557-34-6 | Or similar |
Referencias
- Rupp, W. D., Howard-Flanders, P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. Journal of Molecular Biology. 31 (2), 291-304 (1968).
- Meneghini, R. Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochimica et Biophysica Acta. 425 (4), 419-427 (1976).
- Meneghini, R., Cordeiro-Stone, M., Schumacher, R. I. Size and frequency of gaps in newly synthesized DNA of xeroderma pigmentosum human cells irradiated with ultraviolet light. Biophysical Journal. 33 (1), 81-92 (1981).
- Lopes, M., Foiani, M., Sogo, J. M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Molecular Cell. 21 (1), 15-27 (2006).
- Heller, R. C., Marians, K. J. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 439 (7076), 557-562 (2006).
- Bianchi, J., et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Molecular Cell. 52 (4), 566-573 (2013).
- García-Gómez, S., et al. PrimPol, an archaic primase/polymerase operating in human cells. Molecular Cell. 52 (4), 541-553 (2013).
- Mourón, S., et al. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nature Structural & Molecular Biology. 20 (12), 1383-1389 (2013).
- Wan, L., et al. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Reports. 14 (12), 1104-1112 (2013).
- Keen, B. A., Jozwiakowski, S. K., Bailey, L. J., Bianchi, J., Doherty, A. J. Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Research. 42 (9), 5830-5845 (2014).
- Tirman, S., Cybulla, E., Quinet, A., Meroni, A., Vindigni, A. PRIMPOL ready, set, reprime. Critical Reviews in Biochemistry and Molecular Biology. 56 (1), 17-30 (2021).
- Quinet, A., Lerner, L. K., Martins, D. J., Menck, C. F. M. Filling gaps in translesion DNA synthesis in human cells. Mutation Research, Genetic Toxicology Environmental Mutagenesis. 836, 127-142 (2018).
- Ziv, O., Diamant, N., Shachar, S., Hendel, A., Livneh, Z., Bjergbæk, L. Quantitative measurement of translesion DNA synthesis in mammalian cells. DNA Repair Protocols. Methods in Molecular Biology (Methods and Protocols). 920, (2012).
- Quinet, A., et al. Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Research. 44 (12), 5717-5731 (2016).
- Técher, H., et al. Replication dynamics: biases and robustness of DNA fiber analysis. Journal of Molecular Biology. 425 (23), 4845-4855 (2013).
- Quinet, A., Carvajal-Maldonado, D., Lemacon, D., Vindigni, A. DNA fiber analysis: Mind the gap. Methods in Enzymology. 591, 55-82 (2017).
- Elvers, I., Johansson, F., Groth, P., Erixon, K., Helleday, T. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Research. 39 (16), 7049-7057 (2011).
- Diamant, N., et al. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Research. 40 (1), 170-180 (2012).
- Temviriyanukul, P., et al. Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair. 11 (6), 550-558 (2012).
- Jansen, J. G., et al. Redundancy of mammalian Y family DNA polymerases in cellular responses to genomic DNA lesions induced by ultraviolet light. Nucleic Acids Research. 42 (17), 11071 (2014).
- Quinet, A., et al. Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair. 14 (1), 27-38 (2014).
- Callegari, A. J., Clark, E., Pneuman, A., Kelly, T. J. Postreplication gaps at UV lesions are signals for checkpoint activation. Proceedings of the National Academy of Sciences of the United States of America. 107 (18), 8219-8224 (2010).
- Vogt, V. M. Purification and further properties of single-strand-specific nuclease from Aspergillus oryzae. European Journal of Biochemistry. 33 (1), 192-200 (1973).
- Cordeiro-Stone, M., Schumacher, R. I., Meneghini, R. Structure of the replication fork in ultraviolet light-irradiated human cells. Biophysical Journal. 27 (2), 287-300 (1979).
- Lehmann, A. R., Fuchs, R. P. Gaps and forks in DNA replication: Rediscovering old models. DNA Repair (Amst). 5 (12), 1495-1498 (2006).
- Daigaku, Y., Davies, A. A., Ulrich, H. D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 465 (7300), 951-955 (2010).
- Karras, G. I., Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 141 (2), 255-267 (2010).
- Tirman, S., et al. Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Molecular Cell. 81 (19), 4026-4040 (2021).
- Cong, K., et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Molecular Cell. 81 (15), 3128-3144 (2021).
- Quinet, A., et al. PRIMPOL-mediated adaptive response suppresses replication fork reversal in BRCA-deficient cells. Molecular Cell. 77 (3), 461-474 (2020).
- Jackson, D. A., Pombo, A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. The Journal of Cell Biology. 140 (6), 1285-1295 (1998).
- Schwab, R. A., Niedzwiedz, W. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. Journal of Visualized Experiments: JoVE. (56), e3255 (2011).
- Simoneau, A., Xiong, R., Zou, L. The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes & Development. 35 (17-18), 1271-1289 (2021).
- Taglialatela, A., et al. REV1-Polζ maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Molecular Cell. 81 (19), 4008-4025 (2021).
- Lim, K. S., et al. USP1 Is required for replication fork protection in BRCA1-deficient tumors. Molecular Cell. 72 (6), 925-941 (2018).
- Peng, M., et al. Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Reports. 24 (12), 3251-3261 (2018).
- Huang, J., et al. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Molecular Cell. 52 (3), 434-446 (2013).
- Ding, W., Bishop, M. E., Lyn-Cook, L. E., Davis, K. J., Manjanatha, M. G. In vivo alkaline comet assay and enzyme-modified alkaline comet assay for measuring DNA strand breaks and oxidative DNA damage in rat liver. Journal of Visualized Experiments: JoVE. (111), e53833 (2016).
- Lu, Y., Liu, Y., Yang, C. Evaluating in vitro DNA damage using comet assay. Journal of Visualized Experiments: JoVE. (128), e56450 (2017).
