Representative results obtained with the described method workflow are shown. E15.5 mice were used in the present demonstration, though this protocol is easily adaptable to virtually all embryonic ages ranging from E11 to late E17. In this protocol, either ex utero electroporation (EUE; Figure 2A, 2C-I) or in utero electroporation (IUE; Figure 2B,C, and 2J–Q) were used to deliver plasmids into the progenitor neurons lining the lateral ventricles. These progenitors are the source of future cortical projecting neurons (CPN)15,16. Plasmid mixes were prepared to drive sparse neuron-specific expression of either membrane-targeted (Lyn)-mNeonGreen (Figure 1A) or LifeAct-enhanced (E)GFP (Figure 1B) to evaluate the overall behavior and actin dynamics in growth cones, respectively. Furthermore, a plasmid mix aimed to label individual neurons with either turbo(t)-RFP or zoanthus sp. (Zs) green fluorescent protein (ZsGreen) (Figure 1C) was included. This facilitates the monitoring of growth cone behavior from independent neighboring neurons.
Brain dissection from electroporated embryos is a crucial step that needs to be carefully performed to obtain high-quality slices, preserving the native brain structure. Dissection instruments and vibratome were prepared beforehand and carefully ethanol-sterilized (Figure 3A,B). Next, the heads of electroporated embryos were carefully dissected and the brains were extracted. Here, representative dissection of brains from the embryos subjected to EUE at E15 (Figure 3C–F) and E12.5 (Figure 3G–J) are shown. Brains are immediately encased in an agarose matrix, sliced, and placed on PTFE membrane inserts within a bottom-glass dish for incubation (Figure 3K–M).
The health status of brain slices is a significant point for control to ensure reliable results. A visual inspection for any contamination was performed daily. Additionally, once the culture was finalized, the brain slices are fixed and subjected to immunohistochemistry. Here,4′,6-diamidino-2-phenylindole (DAPI) was used to control the overall cellular organization and vimentin staining to reveal glial organization; particularly, radial glia (RG) scaffold. Typically, successfully cultured brain slices derived from either IUE or EUE show normal cellular distribution as revealed by DAPI and a somewhat organized array of RG with apically oriented pial-contacting processes17 (Figure 4A,B respectively). Occasionally, marked disturbances in the RG scaffolding in cultured brain slices are observed, especially in those derived from EUE electroporation (Figure 4C). Brain slices with extremely disorganized RG scaffold show impaired neuronal migration and defective axon growth (not shown). Hence, controlling the RG scaffold is an easy post-culture method to sort the data obtained from reliable brain slices.
Brain slices derived from either IUE or EUE with Lyn-mNeonGreen-expressing plasmid mix result in similar sparse neuron labeling. A representative pyramidal CPN expressing Lyn-mNeonGreen and the dynamic behavior of its growth cone is shown as an example (Figure 5A and Supplementary Video 1, top left). In addition, neurons were labeled using a plasmid expressing an actin probe to analyze actin dynamics of axonal growth cones in situ (Figure 5B and Supplementary Video 1, bottom left). In situ experiments were also performed with a dual-Cre/Dre fluorophore-expressing plasmid design (Figure 1C and Supplementary Video 1, right). tRFP or ZsGreen fluorophores in this plasmid could be specifically and individually activated by either Dre or Cre recombinases, respectively, in neighboring neurons (Figure 5C). This experimental line-up allows side-by-side analysis of growth cones from control neurons with neighboring modified neurons (any given loss or gain of function). This circumvents variability arising from the use of different slices to test control and experimental conditions.
Kymographs generated from the recorded movie were analyzed, from which dynamic growth parameters such as protrusive activity over time and growth length can be easily obtained (Figure 6A). Note that a simple adjustment in the temporal resolution of the time-lapse allows measurement of axon elongation speed for 2 h (Figure 6A). Furthermore, the variation of growth cone volume over time-a measure of general growth cone dynamic activity-can be easily obtained, in this case with licensed software (Figure 6B and Figure 6E,F). This can be used to evaluate the speed of actin treadmilling and the balance of filopodia/lamellipodia during growth cone exploring activity.

Figure 1: Schemes of the plasmids used in the protocol. (A) pCAG-lox-STOP-lox-Lyn-mNeonGreen. (B) p-Tub-alpha-1-LifeAct-GFP. (C) pCAG-lox-rox-STOP-rox-tRFP-pA-lox-ZsGreen-pA. Relevant information regarding plasmid components and fluorophore's origin is found in the boxes. Please click here to view a larger version of this figure.
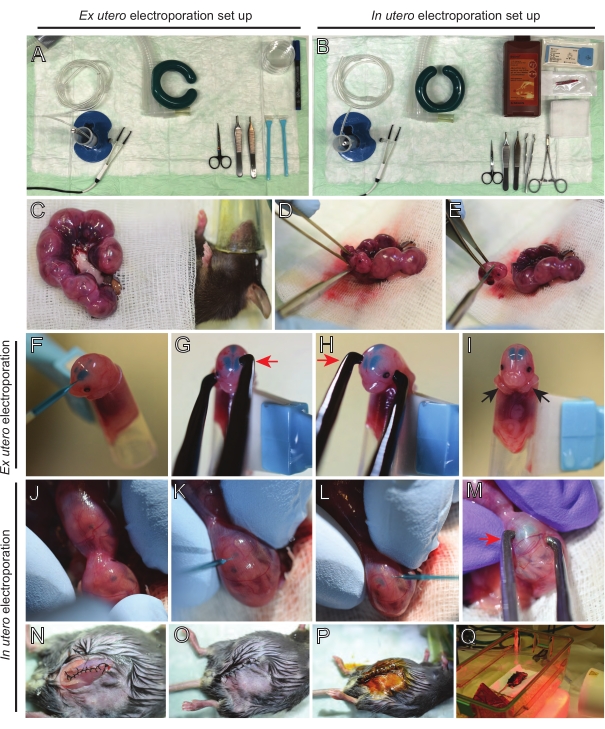
Figure 2: Workflow of ex utero and in utero electroporation of E15.5 mice. (A) Set up of surgery station for ex utero electroporation. (B) Set up of surgery station for in utero electroporation. (C) Uterine horns pulled outside the abdominal cavity of the anesthetized mouse. (D) Extraction of an embryo from the uterine sack. (E) Embryo sacrifice by complete spinal cord transection via a diagonal incision; note that beheading was avoided. (F) Placement of embryo in the holder and injected with DNA/Fast Green mixture into the left lateral ventricle. (G,H) Position the embryo's head between platinum tweezer electrodes with the cathode (red arrow) over the cortex at a 60° angle. (I) Placement of embryo's arms (black arrows) outside the holder to prevent sliding of the embryo during the procedure. (J) Rotation of the embryo inside the uterine sack to expose the head. (K,L) Injection of DNA/Fast Green mixture into embryo's lateral ventricles through the uterine wall. (M) Position the embryo's head between platinum tweezer electrodes with a cathode (red arrow) over the cortex at a 60° angle. (N) Sutured muscle incision via running locking suture. (O) Sutured skin incision via an interrupted suture. (P) Securing of the wound using surgical wound clips and disinfection using betadine. (Q) Placement of the mouse in the recovery cage with far infrared warming light. Please click here to view a larger version of this figure.

Figure 3: Extraction of E15.5 and E12.5 brains and organotypic slice culture procedure. (A) Tools used for the brain extraction procedure. (B) Set up of organotypic culture station. (C–F) Extraction of E15.5 brain. (G–J) Extraction of E12.5 brain. Dotted lines highlight the location of incisions. Red arrows are pointing out the direction of pulling by forceps. (K) Embedding the brain in a 3 cm dish containing 3% low melt agarose, leaving a 1-2 mm agarose spacing gap under the brain. (L) Collection of 150 µm brain slice. (M) Placement of brain slices on PTFE membrane inserts immobilized in a 35 mm dish using paraffin film (blue arrow). Red star marking indicates a given brain slice collection from vibratome (L) and its transfer to PTFE membrane (M). Please click here to view a larger version of this figure.
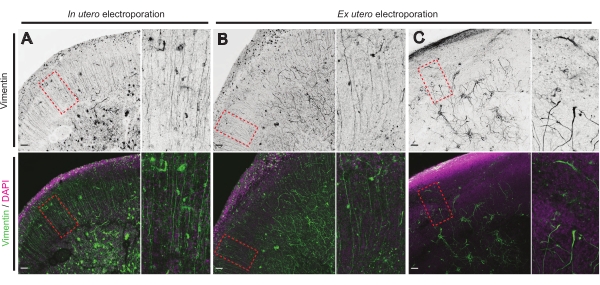
Figure 4: Conserved radial glial cell structure in healthy organotypic slices. Confocal images of E17.5 brain slices revealing RG array (vimentin; green) and overall cell organization (DAPI; magenta) following IUE (A) and EUE (B,C). Note the strong disturbances in the RG array that may occasionally result from EUE (C). Magnifications correspond to the red dotted frames in the main figure: scale bars, 10 µm. Please click here to view a larger version of this figure.
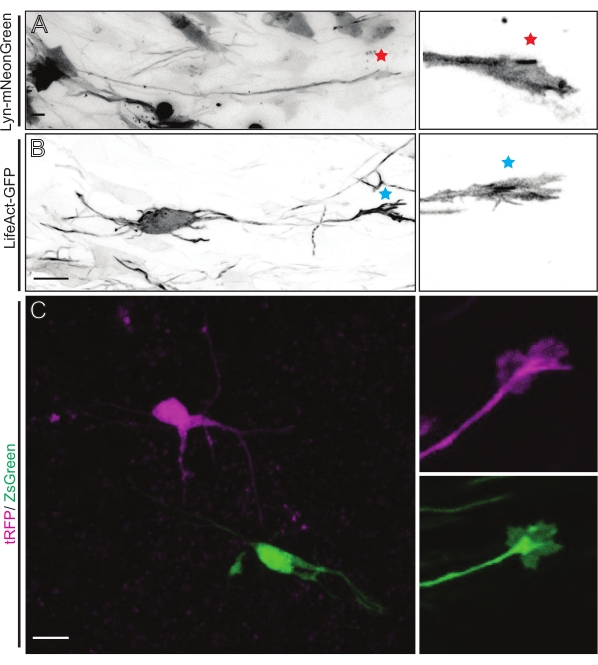
Figure 5: In situ visualization of the growth cone dynamics in acute organotypic slices. (A,B) Neurons and their corresponding growth cones labeled with Lyn-mNeonGreen and LifeAct-GFP, respectively. Red star marking growth cone of Lyn-mNeonGreen expressing neuron. Blue asterisk marking growth cone of LifeAct-GFP expressing neuron. (C) Neighboring neurons labeled with the dual plasmid system containing tRFP (magenta) and ZsGreen (green) and their corresponding growth cones. Growth cones imaged (right) were outside the captured frame (left), obtained shortly after acquiring the growth cone time-lapse; scale bars, 5 µm. Please click here to view a larger version of this figure.
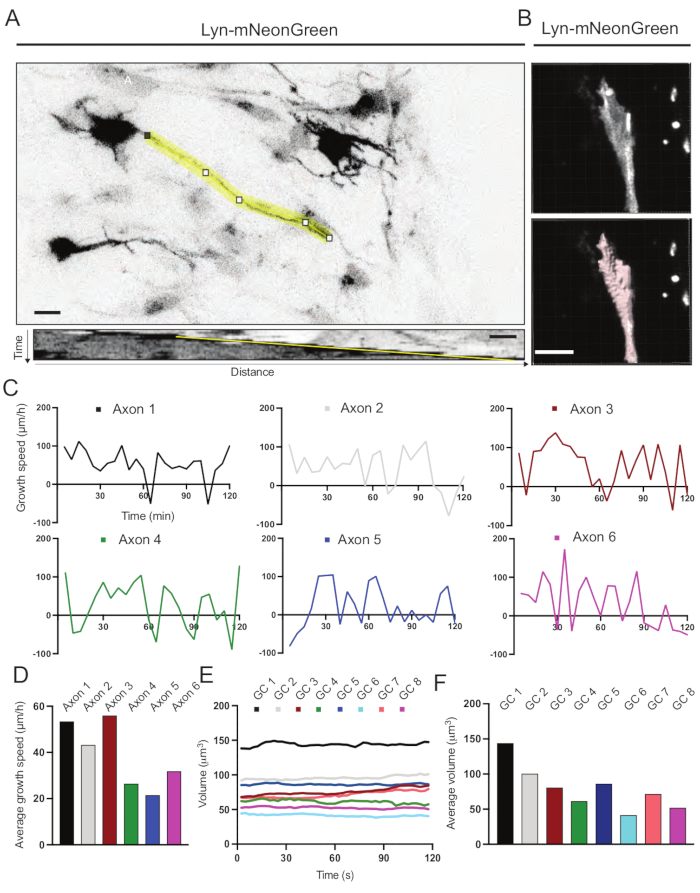
Figure 6: Analysis of axon growth speed and growth cone volume. (A) Axon tracing on a neuron expressing Lyn-mNeonGreen (top) and its corresponding kymograph (below) generated using ImageJ. (B) Reconstruction of z-stack video of growth cone using the image analysis software (top) and the same growth cone highlighted using the surfaces measurement tool (below). (C) Graphs showing changes in growth speed over time for several axons. (D) The average growth speed of axons is quantified in (C). (E) Graph showing the changes in the growth cone volume over time. (F) The average volume of growth cones is quantified in (E); scale bar, 5 µm. Please click here to view a larger version of this figure.
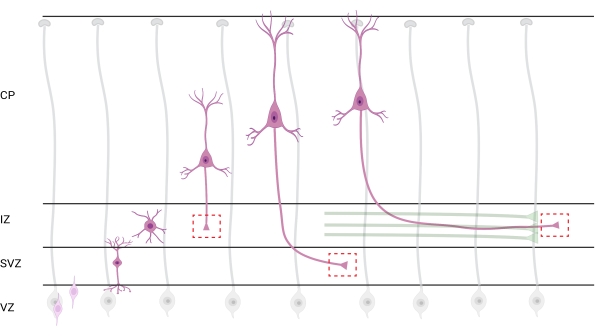
Figure 7: Radial migration and neuronal polarization of pyramidal cortical neurons. Diagram illustrating developing pyramidal cortical neurons (pink) migrating radially from the germinal ventricular zone (VZ) toward the pia surface. Guided by radial glia processes (gray), migrating polarized neurons establish a leading process, future dendrite, and trailing process, future axon, that continue to extend downward toward the intermediate zone (IZ). Dashed red boxes represent the cortical areas where growth cones were imaged. Specifically in the IZ, subventricular zone (SVZ), or joining axon bundles (green). The illustration was created with a web-based tool, BioRender.com. Please click here to view a larger version of this figure.
| Plasmid | Concentration (µg/µL) | Intended use |
| pCAG-lox-STOP-lox-Lyn-mNeonGreen | 0.25 | Labelling of membrane targeted protein (Lyn) |
| + | + | |
| p-Tub-alpha-1-iCre | 0.08 | |
| p-Tub-alpha-1-LifeAct-GFP | 0.125 | Filamentous actin (F-actin) labelling in growth cones |
| pCAG-lox-rox-STOP-rox-tRFP-lox-Lyn-ZsGreen | 1 | Independent labelling of two populations of neighboring neurons |
| + | + | |
| p-Tub-alpha-1-iCre | 0.004 | |
| + | + | |
| p-Tub-alpha-1-Dre | 0.2 |
Table 1: List of plasmids used in the Protocol. Name, concentration, and intended use of each utilized plasmid.
Supplementary Video 1: In situ visualization of the growth cone dynamics in acute organotypic slices. Dynamics of growth cones labeled with Lyn-mNeonGreen (top left) and LifeAct-GFP (bottom left). Neighboring growth cones are differentially labeled with the dual plasmid system containing tRFP (magenta; top right) and ZsGreen (green; bottom right). Imaging interval, 2.5 s. Scale bars, 5 µm. Please click here to download this File.
