Mechanical Separation and Protein Solubilization of the Outer and Inner Perivitelline Sublayers from Hen’s Eggs
Summary
This article reports a simple method to isolate the outer and inner perivitelline sublayers of chicken eggs while minimizing structural alteration and to optimize protein solubilization of each sublayer for proteomic analyses.
Abstract
The perivitelline layer that surrounds the egg yolk plays a fundamental role in fertilization, in egg defense, and in the development of the avian embryo. It is formed by two proteinaceous sublayers that are tightly associated and formed by distinct female reproductive organs. Both structures are assumed to have their own functional specificities, which remain to be defined. To characterize the function of proteins composing each sublayer, the first challenge is to establish the conditions that would allow for the mechanical separation of these two intricate layers, while limiting any structural damage. The second step is to optimize the experimental conditions to facilitate protein solubilization from these two sublayers, for subsequent biochemical analyses. The efficiency of this approach is assessed by analyzing the protein profile of each sublayer by Sodium Dodecyl Sulfate-Poly-Acrylamide Gel Electrophoresis (SDS-PAGE), which is expected to be distinct between the two structures. This two-step procedure remains simple; it requires classical biochemical equipment and reagents; and is compatible with further in-depth proteomics. It may also be transposed to other avian eggs for comparative biology, knowing that the structure and the composition of the perivitelline layer has been shown to have species-specific features. In addition, the non-denaturing conditions developed for sublayers separation (step 1) allow their structural analyses by scanning and transmission electron microscopy. It may also constitute the initial step for subsequent protein purification to analyze their respective biological activities and 3D structure, or to perform further immunohistochemical or functional analyses. Such studies would help to decipher the physiological function of these two sublayers, whose structural and functional integrities are determinant criteria of the reproductive success.
Introduction
The purpose of this method is to provide a protocol that allows subsequent biochemical characterization of the perivitelline layer (PL), a thin proteinaceous layer enclosing the egg yolk and playing a fundamental role in the reproduction of avian species. The PL, also named “vitelline membrane” in the literature, is a three-dimensional network of thick fibers made up of several types of glycoproteins. It consists of an inner perivitelline layer (IPL) (in contact with the yolk) that is assembled in the ovary, and of an outer perivitelline layer (OPL, in contact with the white), lying on the IPL (Figure 1) and produced by the infundibulum. This latter tissue is the funnel-like upper segment of the oviduct receiving the mature yolky follicle after ovulation, and the site where the fertilization takes place. The secretion of OPL occurs after these two events and is followed by the successive deposit of the egg white and the eggshell in other specific segments of the oviduct. The physiological functions of PL are not only related to fertilization and early stages of embryogenesis, but also to the physical and molecular protection of the embryo. The proteomic analysis of chicken egg performed on the whole PL revealed the presence of 137 different proteins1, but the distribution of most of these proteins between IPL and OPL remains to be elucidated. The minimal amount of data available in the literature report that IPL and OPL exhibit very distinct protein profiles2,3,4, which suggests different structural and functional properties. The relative scarcity of data on the distribution of proteins between OPL and IPL is likely due to the difficulty of separating both sublayers that are thin and embedded.
The method presented here describes the conditions to be used to separate the two sublayers with limited impact on their respective histological structure while preserving their protein content, and to provide the protocol permitting the complete solubilization of proteins for subsequent analysis by proteomics. It includes two main steps: 1) PL sampling and OPL/IPL separation and 2) PL, OPL, and IPL treatment for protein solubilization and electrophoresis for mass spectrometry analysis. The workflow is presented in Figure 2. Noticeably, although the present protocol has been optimized for proteomic analyses (Step 2.2), it can be stopped at some steps for histological (e.g., electron microscopy), immunohistochemical analyses, functional studies (step 1), and for purification of salt-soluble proteins in order to characterize their structure and biological activity (step 2.1) (Figure 2).
The first step is the removal of total PL from the egg yolk (Figure 2, step 1.1). All published methods start with the separation of the egg white from the yolk manually or using an egg separator. It is followed by the removal of the remaining egg white and the thick chalazae using forceps or by adsorption on a filter paper5 (Figure 2A). Next, the techniques selected to sample PL are variable depending on the published articles. Some papers include several washes of the yolk, in deionized or distilled water1,5,6, in 0.85 to 1% saline solution2,7,8, in buffering solutions such as 0.15 M NaCl/N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid, pH 7.44, or in 0.01N HCl (pH 2)3. These procedures were applied to chicken, duck, ring-necked pheasant, gray partridge, cockatiel parrot, domestic pigeon, or ratites eggs2,6,9. The removal of PL while the egg yolk is maintained in a Petri dish without aqueous solutions may be very laborious as the PL is fragile and egg yolk tends to stick to PL1,5 and therefore remains difficult to eliminate afterwards. The use of acidic buffer or solution for an hour at 37 °C3 is also not preferred as such conditions may alter PL structure and may result in protein loss. It is also important to remove the area containing the germinal disc as this zone is likely to have a distinct structural and molecular pattern, which does not reflect the whole PL4,6,10 (Figure 2B). This method takes advantage of ideas highlighted by some published articles and proposes some improvements to facilitate PL sampling while preserving its integrity (Figure 2C).
The second step consists of separating the two sublayers (Figure 2, step 1.2). This step is critical as OPL and IPL are tightly bound to each other. This step should be conducted carefully with forceps under a binocular dissecting microscope. Publications reporting OPL/IPL separation are quite limited2,3,7,11 and some of them use specific conditions (acidic buffer at 37 °C for 1 h2,3) that are likely to affect the histological structure of the sublayers and/or contribute to protein loss or yolk or white contaminations. To better distinguish the OPL from the IPL, some authors reported the use of the toluidine blue to slightly color the OPL (the inner layer remains colorless)11. In the method developed, we optimized the conditions so that the separation is easily achieved and does not require the use of any dye (Figure 2D).
The second main step is the solubilization of proteins composing each sublayer. Classically, it is achieved by mixing clean lyophilized1, dried2,6, or freshly prepared3,4,11 PL/sublayers directly with the Laemmli buffer used for electrophoresis. Other authors preferred a preliminary solubilization of layers in 1% NaCl, in a 1% SDS buffering solution2,3,11 or in a solution containing protease inhibitors and SDS6 at room temperature or Triton followed by incubation at 45 °C12, under constant vigorous stirring. Some authors also described a protocol where the PL was incubated in phosphate buffer saline or urea and subjected to sonication13. These treatments were all followed by a centrifugation and dilution in Laemmli buffer and all share the disadvantage of incomplete protein solubilization from layers as the insoluble matter (the pellet obtained after centrifugation) is discarded from the sample to be analyzed.
In addition, the use of denaturing conditions (urea, detergent, high temperature, etc.) by certain authors for protein solubilization is not compatible with subsequent protein purification for characterization as they are likely to irreversibly inactivate proteins, interfere with their biological activities and also impair their 3D structure. For this specific step 2, the protocol includes a first substep allowing for solubilization of the most abundant proteins under non-denaturing conditions after PL/OPL/IPL mechanical de-structuration, which will not interfere with further protein studies, if needed (Figure 2, step 2.1), and a second substep that allows for complete solubilization of proteins for electrophoresis and in-depth proteomics (Figure 2, step 2.2). It combines suggestions taken from various published papers and adjustments resulting from new ideas validated by experimental studies.
Protocol
1. PL, IPL, and OPL samplings
- PL sampling
- Break the freshly laid unfertilized egg manually and use an egg separator lying on a glass beaker to separate the yolk from the white. Remove the chalazae with small scissors and roll the yolk over a filter paper to remove adherent albumen that appears as a transparent and visible structure (Figure 2A).
CAUTION: Be careful not to puncture the PL with scissors. The use of tweezers instead of scissors to remove chalazas may provoke PL breakage and subsequent yolk leakage. The rolling step on the filter paper is critical and should be performed very quickly due to the absorbent properties of the paper filter that may trigger PL breakage. It is also very important to use an unfertilized egg from the day of lay, to optimize the success of this first step and all the subsequent steps. Alternatively, eggs that have been stored less than 10 days in a cooled environment may be used but experimenters must consider potential risks of PL alterations. - Immerse the yolk in a crystallizer containing 10 mM Tris-HCl pH 8 that has been previously cooled down to 4 °C and remove the PL area over the germinal disc within a 1 cm zone using blunt scissors (Figure 2B).
NOTE: Initially, PL was immersed in deionized water, but this approach has some disadvantages, such as the lack of pH control and the difficulties to separate sublayers afterwards (step 1.2). Therefore, several conditions were tested including Tris concentration (Figure 3A) and pH (Figure 3B). The use of a buffer at pH 8, instead of deionized or demineralized water, is in accordance with egg physiology (the pH of egg white is about 7.8 on the day of lay)14 and minimizes protein loss. In contrast, acidic or very alkaline pH is suspected to trigger PL de-structuration and early protein solubilization (Figure 3B). The buffer composed of 10 mM Tris-HCl pH 8 was found to be optimal, as it respects the egg physiology and does not cause significant protein solubilization (protein concentration in the working buffer remains minimal, Figure 3A,B). - Rupture the PL with small scissors in the buffer. Hold the two edges of the ruptured PL with forceps and peel the PL off the yolk.
- Maintain the PL with forceps and rinse the PL several times in several baths of 10 mM Tris-HCl, pH 8 until no trace of yolk is visible. At this stage, ensure that the PL is clean, white, and floating in the buffer. It can be processed in step 1.2 for sublayer separation or directly to step 2.1 for biochemical analyses.
- Break the freshly laid unfertilized egg manually and use an egg separator lying on a glass beaker to separate the yolk from the white. Remove the chalazae with small scissors and roll the yolk over a filter paper to remove adherent albumen that appears as a transparent and visible structure (Figure 2A).
- OPL and IPL sampling
- Spread out the whole PL sample by putting OPL upward into a plastic Petri dish and maintaining it flat with as less wrinkles as possible. Cover the sample with 10 mM Tris-HCl pH 8, 50 mM NaCl. Use a plastic Petri dish (and not glass) to limit PL movements, because PL sticks better to plastic.
- To locate the OPL side, look at where the location of the remaining chalazae that are attached to the OPL and not the IPL. This step requires small NaCl concentration (50 mM) to facilitate sublayer separation.
NOTE: Based on the graph presented in Figure 3C and literature11, this concentration should not lead to major protein loss. Initially, deionized water was used to separate the sublayers, as described previously1,5,6, but this technique was found to be very laborious and resulted in the alteration of the structural PL integrity. Therefore, different NaCl concentrations (Figure 3C) were tested as salt facilitated the separation of the two sublayers. However, it is noteworthy that the use of NaCl concentration >100 mM increases protein solubilization/release from the PL, which is not the objective here (this will be the objective in step 2) (Figure 3C). Adding 50 mM NaCl to the 10 mM Tris-HCl pH 8 buffer facilitates separation of the two sublayers and minimizes protein loss.
- To locate the OPL side, look at where the location of the remaining chalazae that are attached to the OPL and not the IPL. This step requires small NaCl concentration (50 mM) to facilitate sublayer separation.
- Cut the total PL into pieces of about 2 cm x 3 cm with small scissors. Mechanically separate the two layers of each PL piece with ultra-precise tip forceps under a binocular dissecting microscope (Figure 4). Store the resulting IPL and OPL samples individually in microtubes at -80 °C, until further use.
NOTE: The protocol can be paused here. It should be noted that the efficiency and the facility of separation of the two layers greatly depends on the freshness of the egg. Indeed, stored eggs show drastic internal modifications due to the rapid increase in egg white pH and subsequent alteration of the yolk index due to the PL loosening (and weakening). IPL is more fragile as compared with OPL. It is, therefore, preferable to place the face of the OPL above and to separate the two sublayers by pulling on OPL with the forceps. Both sublayers should be handled with precious care.
- Spread out the whole PL sample by putting OPL upward into a plastic Petri dish and maintaining it flat with as less wrinkles as possible. Cover the sample with 10 mM Tris-HCl pH 8, 50 mM NaCl. Use a plastic Petri dish (and not glass) to limit PL movements, because PL sticks better to plastic.
2. Sample treatment for protein solubilization and SDS-PAGE analyses
- Primary protein solubilization
- Freeze-dry IPL and OPL samples individually. Once freeze-drying is completed, cut approximatively 1 mg from each sample in clean microtubes possessing a leak proof screw cap. Keep the remaining samples in tightly closed tubes for prolonged storage at -80 °C.
NOTE: The use of microtubes with a leak proof screw cap ensures hermetical and leak-proof closure to prevent sample rehydration and that will secure the samples. - Mix 1 mg of each lyophilized sublayer with 400 µL of 50 mM Tris pH 7, 500 mM NaCl. Use a mixer-mill for 2 times for 5 min at 30 Hz to disintegrate OPL and OPL structures in microparticles and facilitate protein solubilization. Collect 400 µL of the samples containing the OPL and IPL in two clean microtubes.
NOTE: The buffer used here has been chosen to increase protein solubilization (unlike step 1). This buffer was based on the results presented in Figure 3, which show an increase in protein solubilization at pH 7 (Figure 3B) and using 0.5 M NaCl (Figure 3C). The protocol can be paused here. At this stage, samples can be used for the protein purification of main PL proteins or processed to step 2.2.
- Freeze-dry IPL and OPL samples individually. Once freeze-drying is completed, cut approximatively 1 mg from each sample in clean microtubes possessing a leak proof screw cap. Keep the remaining samples in tightly closed tubes for prolonged storage at -80 °C.
- Protein solubilization for electrophoresis
- Add 5x SDS-PAGE sample buffer (100 µL, 0.25 M Tris-HCl, 0.05% bromophenol blue, 50% glycerol, 5% SDS, 5% beta-mercaptoethanol, pH 6.8) to each 400 µL of the sample and heat at 100 °C for 5 min.
NOTE: The protocol can be paused here, and samples can be stored at -80 °C. At this stage of the protocol, OPL and IPL are completely dissolved. The efficiency of the sample solubilization can be assessed by centrifuging samples for 5 min at 10,000 x g. Complete solubilization is characterized by an absence of visible pellet after centrifugation. Thus, after complete solubilization, it is considered here that 1 mg of lyophilized sublayer corresponds to 1 mg of proteins (the protein concentration in the 500 µL sample is 2 mg/mL). Indeed, PL is composed of more than 80% protein2,15. The use of a 5x sample buffer limits sample dilution but a 1x or 2x sample buffer can also be used. - Load a maximum of 20 µg of proteins/lane on a SDS polyacrylamide gel (4%–20% gradient gel) and perform electrophoresis at 120 V using a vertical electrophoresis system.
NOTE: The use of a 4%–20% gradient gel is not mandatory but is preferable here as OPL and IPL exhibit proteins with molecular weights ranging from <10 kDa to >250 kDa. It might be also useful to keep the stacking gel (that is usually removed), as the presence of insoluble proteins or protein aggregates at the bottom of wells would indicate poor solubilization and a possible missing step during the sample preparation. - After electrophoresis, remove gels from the glass plates and stain with Coomassie Brilliant Blue solution (50% H2O, 40% EtOH, 10% acetic acid, and R250 Coomassie Brilliant Blue) for 30 min.
- De-stain with a solution consisting of 50% H2O, 40% EtOH, 10% acetic acid solution until the gel background appears light blue.
- Finally transfer the gel in Petri dishes containing deionized water, to achieve de-staining and rehydration of polyacrylamide gels. Proteins should appear as blue bands of a transparent background.
- Add 5x SDS-PAGE sample buffer (100 µL, 0.25 M Tris-HCl, 0.05% bromophenol blue, 50% glycerol, 5% SDS, 5% beta-mercaptoethanol, pH 6.8) to each 400 µL of the sample and heat at 100 °C for 5 min.
Representative Results
OPL and IPL were separated from the PL of a freshly laid unfertilized egg to analyze their protein profiles by SDS-PAGE. The experimental workflow of the whole protocol is illustrated in Figure 2. Figure 3 shows protein release at different Tris concentrations (Figure 3A), various pH (Figure 3B) and different NaCl concentrations (Figure 3C). The resulting two sublayers were observed under the light of the binocular dissecting microscope and are shown in Figure 4 where IPL is translucent while OPL is dense, cloudy, and whitish. These features can partly testify for the efficiency of the sublayer separation.
Following the separation, OPL and IPL were lyophilized independently, and their protein content was completely dissolved thanks to a combination of mechanical grinding, the use of an anionic detergent (SDS), and a reducing agent (beta-mercaptoethanol), followed by boiling. After migration in a polyacrylamide gel (SDS-PAGE electrophoresis), OPL and IPL samples exhibit distinct electrophoretic profiles (Figure 5), as expected.
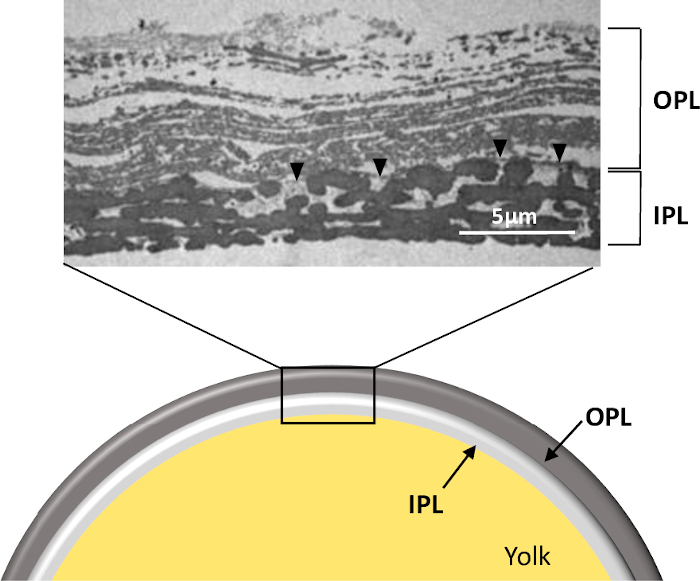
Figure 1: Structure of the PL of a freshly laid unfertilized egg. Schematic representation and transmission electron microscopy micrograph of the whole PL in cross-section (personal data, ©Plateforme IBiSA de Microscopie Electronique, University and CHRU of Tours, France, S. Georgeault). Note that this picture was obtained from PL prepared as presented in this protocol. OPL, outer perivitelline layer; IPL, inner perivitelline layer. Black arrowhead indicates the continuous membrane separating the two sublayers. Please click here to view a larger version of this figure.
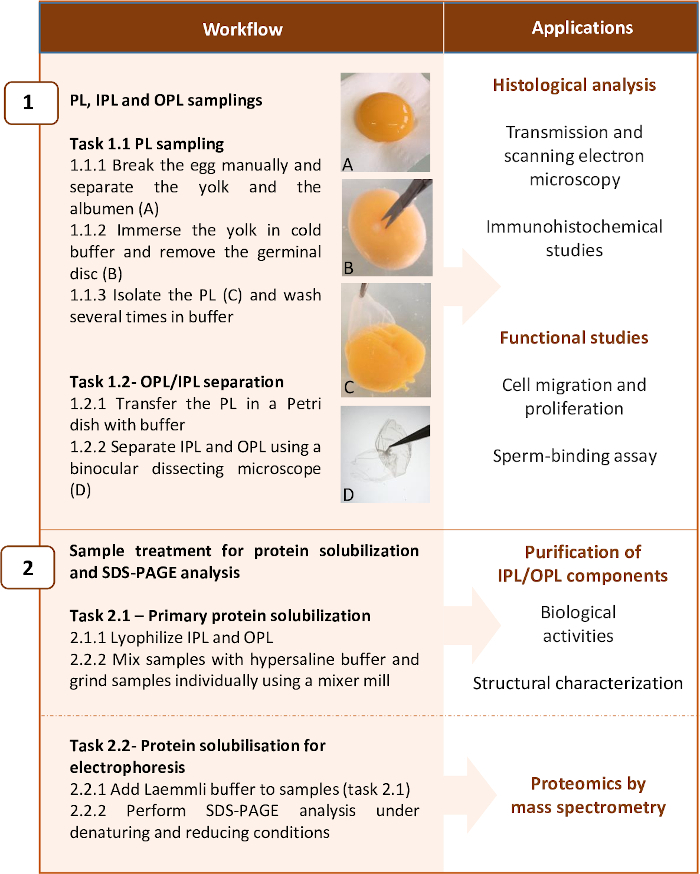
Figure 2: Workflow summarizing the two main steps of the protocol and examples of applications. The protocol has been optimized for proteomic analysis of PL layers, but it can be stopped at some steps for other applications. Please click here to view a larger version of this figure.
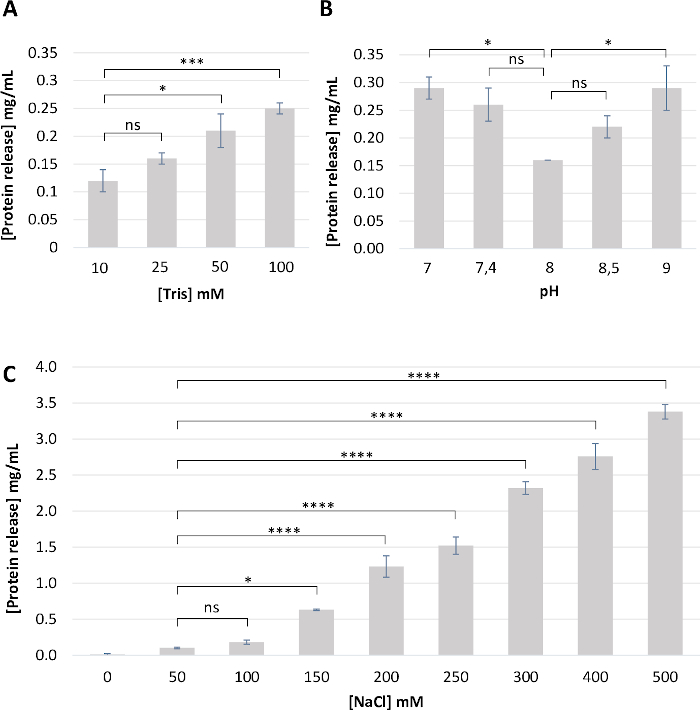
Figure 3: Optimization of the buffer composition for PL sampling. Briefly, clean PL (n = 4) was incubated in the same volume of the various buffering solutions for 2 h. PL was removed, and the protein concentration was estimated by measuring absorbance at 280 nm in the remaining buffer. (A) Effect of Tris-HCl concentration at pH 8. (B) Effect of pH (7 to 9). (C) Effect of NaCl concentration. From these results, we concluded that the buffer composed of 10 mM Tris-HCl, pH 8, 50 mM NaCl is the most appropriate as it limits protein loss. Results are expressed as a mean ± standard deviation of four different PL samples. Statistical analyses were performed using one-way ANOVA followed by a Tukey’s multiple comparisons test. P-value <0.05 was considered significant (ns, not significant; * p<0.05, *** p<0.001, **** p<0.0001). Please click here to view a larger version of this figure.
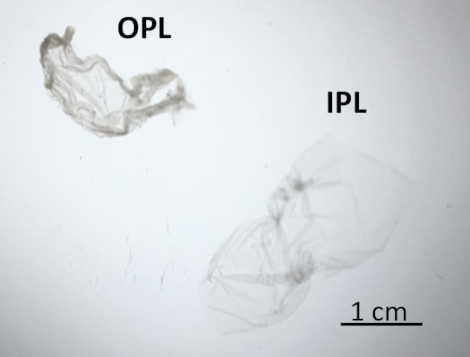
Figure 4: OPL and IPL features under a binocular dissecting microscope. After separation, IPL (on the right) appeared translucent and OPL (on the left) was opaque. Please click here to view a larger version of this figure.
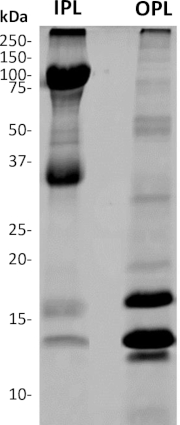
Figure 5: SDS-PAGE analysis of separated OPL and IPL from freshly laid unfertilized egg. 4%–20% acrylamide gel followed by Coomassie brilliant blue staining. The mass of molecular weight standard bands is indicated in kDa. The OPL profile mainly composed of proteins with a molecular mass >30 kDa while the major bands detected on the IPL profile have molecular masses <30 kDa. Please click here to view a larger version of this figure.
Discussion
The success of the present protocol relies on two critical steps that were independently optimized: the mechanical separation of OPL and IPL that need to be as less de-structuring as possible, followed by the complete protein solubilization of each sublayer, which is, conversely, de-structuring and denaturing. It also highlights some specific points that need to be taken into consideration to ensure the successful achievement of each step.
The protocol does not depend on the eggshell color, the egg weight, the type of production, or the genetic strain of the hen. However, it depends on the freshness of the egg. Indeed, the egg undergoes profound internal physicochemical modifications quickly after being laid, although these changes can be delayed when storage is performed under refrigerated conditions14,15. The loss of carbon dioxide through the eggshell pores during storage results in the increase of egg white pH (from 7.8 to 9.5) while some water exchanges occur between the yolk and the white, across the PL. Both modifications negatively impact the molecular and histological structure of the PL that becomes weakened and less tense14,15, likely due to physicochemical modifications of its protein content16,17,18. It is assumed that the separation of sublayers will become very difficult with stored eggs especially if the storage is conducted for a long period at room temperature. Up to date, the protocol has been performed using eggs stored for less than 4 days at 4 °C and the maximal duration of storage for an egg to efficiently sample PL sublayers is not yet known. In addition, if the objective is the analysis of each sublayer, it is crucial to use unfertilized eggs. On the day of lay, the embryo of a fertilized egg is 23 h old and its development is very rapid afterwards, if the egg is incubated. Indeed, embryonic development and the growth of some extraembryonic structures expand using PL as a substratum, which is thus rapidly degraded, and replaced by the extraembryonic yolk sac19.
After manual separation of the yolk and the egg white, the yolk needs to be cleaned from egg white traces and from chalazae that remained tightly attached to the PL. The tip is to roll the yolk carefully onto on a filter paper and to perform extensive washes of the yolk in buffered solutions (10 mM Tris-HCl pH 8). The pH used for the buffer is consistent with the physiological pH of the egg white14 and has been shown to minimize protein loss (Figure 3). The use of a refrigerated buffer will then help to remove the PL from the yolk because at 4 °C, the buffer will facilitate PL removal as the lipidic yolk retracts and becomes less fluid at this temperature. Additional washes will allow for the removal of yolk residues from the PL. It is also important to cut out the zone corresponding to the germinal disc because this structure that contains female pronucleus may not be representative of the PL. It is the site of fertilization and multiplication of the embryonic cells when the egg is fertilized. It is supposed to have a particular composition that may be not representative of the whole PL, which is the reason why it was removed. This specific step is strongly facilitated when the yolk is immersed in a cold buffer. Although this germinal disc structure is discarded in the present protocol (out of the objectives), specific analyses of this area in fertilized eggs could be very useful for scientists interested in studying the evolution of the PL content (proteomics and activity) and structure (electron microscopy), during the early stages of embryogenesis19,20,21. At this stage, the whole PL is structurally intact and can be processed for further structural analysis by electron microscopy (Figure 2), to step 1.2 or to step 2.1 (if the objective is to analyze the whole PL).
Step 1.2 needs to be conducted under a binocular dissecting microscope, as the PL is very thin and fragile (about 10 µm thick). The separation of sublayers is nearly impossible in water or in buffer lacking minimal salt, and initial attempts using these options have resulted in serious PL damage. Thus, the buffer selected for this step remains at physiological pH (pH 8) and contain 50 mM NaCl. This step is arduous and must be performed carefully and gently to prevent PL tearing. Once separated, the two sublayers can be easily identified as they exhibit peculiar physical features (opaque versus translucent). A lack of homogeneity of the IPL under the light of the microscope should reflect an incomplete separation and thus the presence of the remaining spots of OPL present on IPL. In addition, it is very difficult to treat the whole membrane and the use of 2 cm x 3 cm pieces greatly enhance the chances of success. The resulting sublayer obtained is suitable for histological study by electron microscopy (Figure 1). It is noteworthy that a specific linear structure (0.05 to 1 μm thick) named the continuous membrane (CM) is visible on the micrograph (Figure 1) and remains usually attached to one or the other sublayer during the process of separation. It has been previously published that when using a relatively high salt molarity for separation (500 mM), the CM remained attached to OPL7 while the use of water5 will favor the attachment of CM to IPL. In this protocol, a low salt molarity is used to achieve the specific objectives (PL sublayer structurally intact and minimal loss of proteins), which means that this CM is likely associated to IPL. However, additional analysis of IPL and OPL by transmission electron microscopy would clearly state on this hypothesis. PL, OPL, or IPL layers obtained at the end of step 1 may also be used for in vitro assays for sperm-egg binding analyses22 or to analyze the early steps of embryonic development23,24.
Step 2 refers to the solubilization of protein content, partially (step 2.1) or completely (step 2.2). PL and/or OPL are first lyophilized to remove water molecules and to allow for precise weight measurements. Indeed, PL is mainly composed of water (88%)7, while the dry matter contains essentially proteins (80% to 90% depending on studies), carbohydrates, fats, and minerals2,7,25. Considering that the water contained in PL is removed by freeze-drying, that carbohydrates are essentially recovered in PL glycoproteins, and that fat and minerals originate from yolk are essentially discarded by extensive washes, it is assumed that the resulting PL is composed almost exclusively of proteins. Thus, the weight value obtained after complete lyophilization should mainly correspond to proteins. Equal amount of PL, OPL, and/or IPL are then diluted in the buffer containing 0.5 M NaCl. The high salt molarity combined to the mechanical destruction of the fibrous network by grinding greatly facilitates protein solubilization under non-denaturing conditions. This step is followed by the complete solubilization using boiling, SDS detergent and the reducing agent beta-mercaptoethanol (step 2.2). Indeed, some IPL proteins are SDS-soluble glycoproteins25,26,27 and the major constituents of OPL are NaCl-soluble1,11. Using this protocol, we did not see any remaining pellet of insoluble proteins after centrifugation, which indicates that the solubilization was likely complete. Proteins from PL, OPL, and/or IPL were then analyzed by SDS-PAGE. Resulting protein profiles are consistent with published literature2,4,11,16, but the resolution and intensity of the signal is highly improved and much more homogenous between various IPL and OPL independent samplings.
In addition, quantitative comparison between OPL and IPL is made possible thanks to the accuracy of the weight that we inferred for the respective sublayers2,7. Moreover, both OPL and IPL profiles are very distinct and except a faint band at 14 kDa and 75 kDa, both PL sublayers do not visibly share bands displaying the same molecular weight. This observation corroborates the efficiency of sublayers separation with no significant cross-contamination. According to the only PL proteomic analysis published1, the most intense bands in OPL (<30 kDa) should correspond to lysozyme (14 kDa), vitelline membrane outer layer protein 1 (17 kDa), avian beta-defensin 11 (apparent molecular weight of 12 kDa), while the intense bands of IPL (>30 kDa) are likely Zona-Pellucida protein 1 (102 kDa) and Zona-Pellucida protein 3 (47 kDa). The distribution of the very abundant PL proteins ovotransferrin (78 kDa), ovalbumin-related protein X (45 kDa), ovalbumin (43 kDa)1 between sublayers remains to be elucidated. Indeed, the high molecular weight of these proteins (>30 kDa) suggests that they are IPL proteins based on the SDS-PAGE profile, but their tissue-specificity (oviduct-expressed proteins) would rather associate these proteins to OPL28,29. Moreover, some have suggested a potential contamination of PL samples by egg white and egg yolk proteins due to insufficient washings1. This lacking information is the basis for the development of the present protocol, which will be further used for in-depth quantitative proteomics of PL, OPL, and IPL.
Alternatively, from step 2.1 and after centrifugation to remove the insoluble matter, it is possible to extract some specific proteins for further biochemical and biological characterization. Such an approach has been recently applied to characterize the biological activities and/or the 3D structure of the two main components of the OPL, the vitelline membrane outer layer protein 1 (VMO1) and avian beta-defensin 11 (AvBD11)30,31. The availability of these proteins as purified molecules may also help to produce specific antibodies for additional experiments such as immunohistochemistry or for the development of quantitative assays, including Enzyme-Linked Immunosorbent Assays.
The two major limitations of this protocol are (1) the challenge with sublayers separation especially if eggs have been stored more than 4 days at 4°C and (2) the fact that the complete protein solubilization requires reducing and denaturing buffers, which impair the intrinsic biological activity of the resulting samples. Regarding the first limitation, mechanical separation requires technical skills but is easily achieved after 2 or 3 experimental trainings. Some IPL small pieces may remain attached to the OPL and vice versa as both sublayers are tightly associated. However, contamination of one sublayer by the other should be minimal if the mechanical separation is performed cautiously. Typical IPL and OPL SDS-PAGE profiles after optimal sublayer separation are illustrated in Figure 5. Regarding the second limitation, experimental steps presented in this article have been optimized for proteomic analyses, in order to provide an exhaustive list of proteins composing each sublayer. For further characterization of the biological activities of each protein, it is necessary to stop at step 2.1.2, before using denaturing conditions (step 2.2.1, use of buffer with SDS and beta-mercaptoethanol followed by boiling). However, it is noteworthy that proteins resulting from step 2.1.2 are only salt-soluble proteins while salt-insoluble proteins form aggregates. One option to further assess their respective activity may be to produce salt-insoluble proteins as recombinant proteins using heterologous systems (E. coli, baculovirus, yeast, etc.).
This protocol may be adapted to other avian eggs for comparative studies to better appreciate the originality of each species. Indeed, PL displays some bird-specificities both structurally and in terms of protein composition, likely due to adaption to new environments but also to developmental specificities2,6,9,32. The development of this protocol that requires moderate technicity and classical equipment/materials opens new research avenues in the field of bird reproduction and speciation studies.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
We are grateful to Philippe Didier and Karine Anger (INRAE, PEAT, 37380, Nouzilly, France, https://doi.org/10.15454/1.5572326250887292E12) for providing unfertilized Isa-brown eggs. We also thank University of Tours, France for financing M. Bregeon’s PhD. This work received a financial support from The French National Research Agency (EQLIPSE, ANR-19-CE21-0006).
Materials
| Binocular dissecting microscope | Vision Engineering, France | Model Mantis Elite | |
| Lyophilizer | Cryotec, France | Model Cosmos 80 | |
| Mini-Protean II electrophoresis cell | Biorad, Hercules, USA | 1652960 | Any apparatus adapted for protein electrophoresis |
| Mixer Mill MM400 | Retsch, Hann, Germany | 20.745.0001 | Mixer mill adapted for for dry, wet, and cryogenic grinding of small amounts of sample |
| Ultra fine dissection scissors in stainless steel length 12 cm | Dutscher, Brumath | 5066 | |
| Ultra precise tip forceps anti-magnetic stainless steel 9.5x 109.3 mm | Dutscher, Brumath | 327005 |
Referencias
- Mann, K. Proteomic analysis of the chicken egg vitelline membrane. Proteomics. 8 (11), 2322-2332 (2008).
- Chung, W. H., Lai, K. M., Hsu, K. -. C. Comparative study on histological structures of the vitelline membrane of hen and duck egg observed by cryo-scanning electron microscopy. Journal of Agricultural and Food Chemistry. 58 (3), 1794-1799 (2010).
- Kido, S., Doi, Y. Separation and Properties of the Inner and Outer Layers of the Vitelline Membrane of Hen’s Eggs. Poultry Science. 67 (3), 476-486 (1988).
- Steele, M. G., Meldrum, W., Brillard, J. P., Wishart, G. J. The interaction of avian spermatozoa with the perivitelline layer in-vitro and in-vivo. Journal of Reproduction and Fertility. 101 (3), 599-603 (1994).
- Bain, J. M., Hall, J. M. Observations on the development and structure of the vitelline membrane of the hen’s egg: an electron microscope study. Australian Journal of Biological Sciences. 22 (3), 653-665 (1969).
- Damaziak, K., Kieliszek, M., Buclaw, M. Characterization of structure and protein of vitelline membranes of precocial (ring-necked pheasant, gray partridge) and superaltricial (cockatiel parrot, domestic pigeon) birds. PLoS One. 15 (1), 0228310 (2020).
- Bellairs, R., Harkness, M., Harkness, R. D. The vitelline membrane of the hen’s egg: a chemical and electron microscopical study. Journal of Ultrastructure Research. 8 (3-4), 339-359 (1963).
- Kido, S., et al. Characterization of vitelline membrane outer layer protein I, VMO-I: amino acid sequence and structural stability. Journal of Biochemistry. 117 (6), 1183-1191 (1995).
- Damaziak, K., et al. Comparative analysis of structure and strength of vitelline membrane and physical parameters of yolk of ostrich, emu, and greater rhea eggs. Poultry Science. 97 (3), 1032-1040 (2018).
- Kirunda, D. F., McKee, S. R. Relating quality characteristics of aged eggs and fresh eggs to vitelline membrane strength as determined by a texture analyzer. Poultry Science. 79 (8), 1189-1193 (2000).
- Back, J. F., Bain, J. M., Vadehra, D. V., Burley, R. W. Proteins of the outer layer of the vitelline membrane of hen’s eggs. Biochimica Et Biophysica Acta. 705 (1), 12-19 (1982).
- Waclawek, M., Foisner, R., Nimpf, J., Schneider, W. J. The chicken homologue of zona pellucida protein-3 is synthesized by granulosa cells. Biology of Reproduction. 59 (5), 1230-1239 (1998).
- Okumura, H., et al. A newly identified zona pellucida glycoprotein, ZPD, and dimeric ZP1 of chicken egg envelope are involved in sperm activation on sperm-egg interaction. Biochemical Journal. 384, 191-199 (2004).
- Guyot, N., Rehault-Godbert, S., Nys, Y., Baron, F., Roberts, J. Understanding the natural antibacterial defences of egg white and their regulation. Achieving Sustainable Production of Eggs Volume 1. 1, 161-193 (2016).
- Trziszka, T., Smolinska, T. Chemical Characterization of the Vitelline Membrane of Hens Eggs. Food Chemistry. 8 (1), 61-70 (1982).
- Berardinelli, A., Ragni, L., Giunchi, A., Gradari, P., Guarnieri, A. Physical-mechanical modifications of eggs for food-processing during storage. Poultry Science. 87 (10), 2117-2125 (2008).
- Kelley, A. J. The effects of storage time on vitelline membrane protein banding patterns and interior egg quality of eggs from nonmolted and molted hens. Texas A&M University. , (2003).
- Schafer, A., Drewes, W., Schwagele, F. Analysis of vitelline membrane proteins of fresh and stored eggs via HPLC. Zeitschrift Für LebensmittelUntersuchung Und-Forschung A. Food Research and Technology. 206 (5), 329-332 (1998).
- Romanoff, A. L. . The avian embryo: Structural and functional development. , (1960).
- Haas, H. J., Spratt, N. T. Contributions to an analysis of the avian vitelline membrane’s potential to promote outgrowth of the yolk sac-serosal membrane. The Anatomical Record. 184 (2), 227-231 (1976).
- Jensen, C. Ultrastructural changes in the avian vitelline membrane during embryonic development. Journal of Embryology and Experimental Morphology. 21 (3), 467-484 (1969).
- Barbato, G. F., Cramer, P. G., Hammerstedt, R. H. A practical in vitro sperm-egg binding assay that detects subfertile males. Biology of Reproduction. 58 (3), 686-699 (1998).
- New, D. A. T. The Adhesive Properties and Expansion of the Chick Blastoderm. Journal of Embryology and Experimental Morphology. 7 (2), 146-164 (1959).
- New, D. A. T. A New Technique for the Cultivation of the Chick Embryo. Journal of Embryology and Experimental Morphology. 3 (4), 326-331 (1955).
- Kido, S., Janado, M., Nunoura, H. Macromolecular components of the vitelline membrane of hen’s egg. I. Membrane structure and its deterioration with age. Journal of Biochemistry. 78 (2), 261-268 (1975).
- Kido, S., Janado, M., Nunoura, H. Macromolecular components of the vitelline membrane of hen’s egg. II. Physicochemical properties of glycoprotein I. Journal of Biochemistry. 79 (6), 1351-1356 (1976).
- Kido, S., Janado, M., Nunoura, H. Macromolecular components of the vitelline membrane of hen’s egg. III. Physicochemical properties of glycoprotein II. Journal of Biochemistry. 81 (5), 1543-1548 (1977).
- Réhault-Godbert, S., et al. Ovalbumin-related protein X is a heparin-binding ov-serpin exhibiting antimicrobial activities. Journal of Biological Chemistry. 288 (24), 17285-17295 (2013).
- Gautron, J., et al. Ovotransferrin is a matrix protein of the hen eggshell membranes and basal calcified layer. Connective Tissue Research. 42 (4), 255-267 (2001).
- Guyot, N., et al. Proteomic analysis of egg white heparin-binding proteins: towards the identification of natural antibacterial molecules. Scientific Reports. 6, 27974 (2016).
- Guyot, N., et al. function, and evolution of Gga-AvBD11, the archetype of the structural avian-double-beta-defensin family. Proceedings of the National Academy of Sciences of the United States of America. 117 (1), 337-345 (2020).
- Mori, M., Masuda, N. Proteins of the vitelline membrane of quail (Coturnix coturnix japonica) eggs. Poultry Science. 72 (8), 1566-1572 (1993).
