The principle of the fluorescence leakage assay is shown in the Figure 1. In detail, large unilamellar vesicles (LUVs) encapsulating a fluorescent dye and a quencher (no fluorescence signal) are treated with the biomolecule of interest. Due to the interaction of the peptide with lipid membranes, which could imply membrane permeability, reorganization or even rupture, the fluorescence dye and the quencher are released from the LUVs. Subsequent dilutions in the buffer results in an increased fluorescence signal.
Although this scheme displays a test with free peptides, the advantage of the system lies in the ability to also test cargo-conjugated peptides, peptide-based nanoparticles or other biopolymers, which are suspected to destabilize lipid membranes. Although a preliminary optimization of the protocol especially with regards to the molecules tested is required, this fluorescence leakage assay might be extended to a huge variety of membrane-interacting components. In the present protocol, we show some results obtained with the CPPs and their complexed (non-covalent strategy) or conjugated (covalent strategy) forms. The following examples imply WRAP alone as well as siRNA-loaded WRAP-based nanoparticles and two different peptide-conjugates (WRAP-iCAL3617 and Penetratin-iCAL36).
With regard to non-covalent strategy, fluorescence leakage assay with peptides and with their corresponding siRNA-loaded nanoparticles in the presence of LUVs are displayed in Figure 2. The vesicles are composed of a mixture of DOPC/SM/Chol (4:4:2) reflecting the plasma membrane as described in Konate et al.21 and are usually used to directly evaluate the possibility of lipid bilayer interaction and/or transduction properties of free WRAP and WRAP-based nanoparticles7. In the absence of peptides, no leakage is observed (baseline during the first 100 s). Addition of free WRAP on the LUVs induces a significant increase of fluorescence revealing an important LUV leakage and ANTS release. After 15 min, a leakage of 67.8% ± 0.4% compared to the Triton condition (positive control) is obtained at the used concentration (2.5 µM) of WRAP peptide. It should be noted here that several different concentrations have also been tested and revealed a dose-dependent fluorescence increase, corresponding to a dose-dependent LUV leakage7. In contrast, when WRAP is assembled at the same concentration with siRNA to form peptide-based nanoparticles, the leakage is 1.5-fold weaker (40.5% ± 0.5%) compared to the free peptide (Figure 2). Similar leakage values have been reported for the RICK peptide (60%) or the RICK:siRNA nanoparticles (28%)22. The difference in values between free peptide compared to nanoparticles might be explained by the fact that, when engaged in the nanoparticles, a substantial part of the peptide is involved in direct interactions with the siRNA, reducing the peptide availability for interactions with lipids.
Concerning the covalent strategy, fluorescence leakage assays with CPP-conjugates in the presence of LUVs are shown in Figure 3. With the same LUV composition [DOPC/SM/Chol (4:4:2)], two conjugated peptides are applied: WRAP-iCAL36 and Penetratin-iCAL36. As previously noticed, no leakage is observed in the absence of peptides. 15 min after injection of 2.5 µM of Penetratin-iCAL36, no significant fluorescence increase is detected (2.3% ± 0.7%), whereas an addition of 2.5 µM of WRAP-iCAL36 induces a net leakage characterized by very strong fluorescence signal (85.8% ± 11.1%)17. These observations indicate that for some peptides, or conjugates, no fluorescence leakage might occur, suggesting no peptide/membrane interactions or no lipid bilayer disturbance. This is in accordance with previously published results showing that Penetratin as well as Tat were not able to destabilize membranes23,24,25. It should be highlighted that the membrane destabilization properties of a CPP could change depending on the coupled cargo26.
Furthermore, although WRAP-iCAL36 caused a strong leakage, additional studies do not reveal specific cellular internalization, indicating that these conjugates remained inside the lipids bilayer of the plasma membrane17. In contrast to the WRAP nanoparticle, we suppose that the WRAP-cargo conjugate is able to destabilize the LUV membrane by sticking between the lipid chains or by forming pores.
These results indicate that the fluorescence leakage assay might reveal the ability of some CPPs to develop peptide/membrane interactions, which could lead to a more or less pronounced membrane permeability. Moreover, these interactions might occur whatever the strategy of cargoes delivery (nanoparticles versus conjugates). Conversely, this method does not discriminate whether a CPP, which does not induce any fluorescence leakage, might still interact with the bilayer or biological membrane of the lipids. This kind of behavior requires additional approaches such as zeta-potential measurements, FRET between peptide and membrane, or tryptophan fluorescence experiments to mention only a few examples.
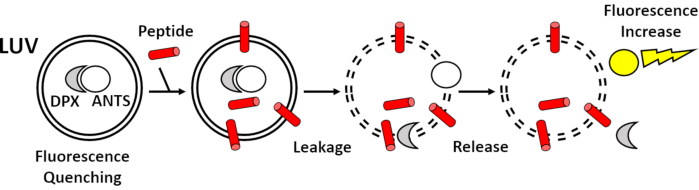
Figure 1: Principle of the fluorescence leakage assay. LUVs were loaded with a fluorescent dye (ANTS) in white and its corresponding quencher (DPX) in grey. In the absence of peptides (in red), no fluorescence signal was observed because ANTS fluorescence was quenched by DPX. Addition of peptides on the LUVs induced membrane permeability and the subsequent release of both ANTS and DPX resulting in a significant increase in ANTS fluorescence (yellow). Please click here to view a larger version of this figure.
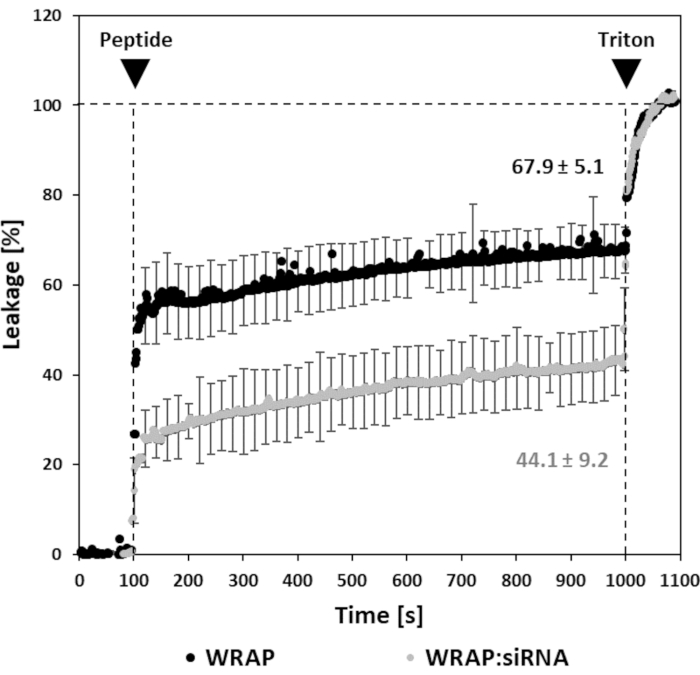
Figure 2: Fluorescence leakage assay with free WRAP and WRAP:siRNA nanoparticles. Peptide alone and siRNA-loaded nanoparticles were applied on LUVs at the peptide concentration of 2.5 µM. WRAP-based nanoparticles were formulated at a peptide:siRNA molar ratio of 20 (R 20) as described in Konate et al.7,16. Black arrows show injections of peptide (nanoparticle) and Triton X-100, respectively. Please click here to view a larger version of this figure.
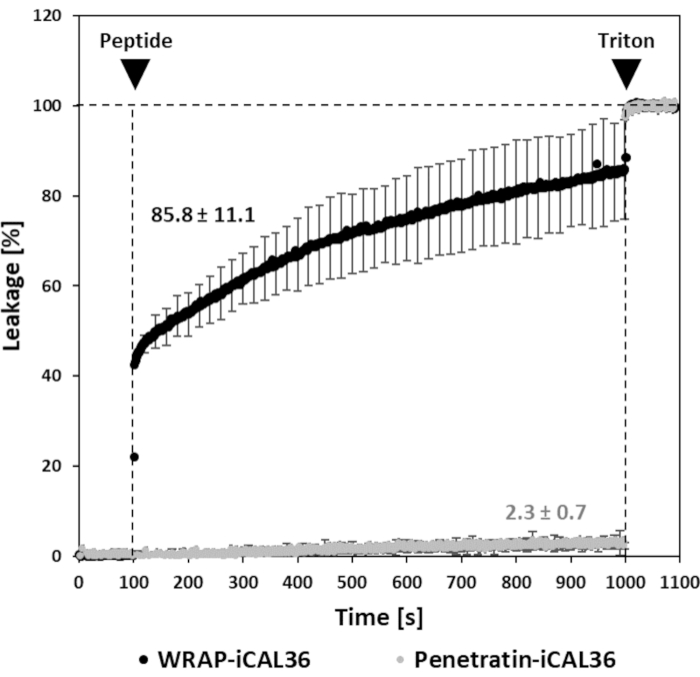
Figure 3: Fluorescence leakage assay with WRAP-iCAL36 and Penetratin-iCAL36 conjugates. Conjugates were applied on LUVs at the concentration of 2.5 µM. Black arrows show injections of peptide and Triton X-100, respectively. Please click here to view a larger version of this figure.
