1. Gel preparation
- Silanization of the gel support
- Activate the coverslip or glass-bottom Petri dish (which will be used as gel support) with a UV lamp for 2 min (wait 30 s before exposure to the UV lamp to avoid exposure to residual ozone).
- Silanize the coverslip/glass-bottom dish using 200 µL aminopropyltrimethoxysilane (APTMS) for 5 min. This will prepare the support for the covalent binding of the gel.
- Thoroughly wash the coverslip/glass-bottom dish with ultra-pure water.
- Dry the coverslip/glass-bottom dish using vacuum aspiration.
- Preparation of the 18mm coverslip used to flatten the gel
- To prepare the coverslips, first put them into a ceramic coverslip holder. Then put the coverslip holder into a small beaker (50 mL) and pour siliconizing reagent (stored at 4 °C, reusable) over the coverslips, being sure to completely cover them.
- Cover the beaker with aluminum foil and incubate for 3 min at room temperature. While waiting, fill a large beaker (500 mL) with ultra-pure water. After 3 min of incubation in siliconizing reagent, transfer the coverslip holder with coverslips to the beaker of water.
- Thoroughly rinse the coverslips with ultra-pure water, dry them well and keep on paper wipes. For best results, proceed immediately to the next section.
- Gel polymerization
- For gels of 0.5 kPa, mix 75 µL of 40% acrylamide with 30 µL of 2% bisacrylamide (crosslinker) and 895 µL of phosphate-buffered saline (PBS). This premix can be stored for up to one month at 4 °C.
- To 167 µL of 0.5 kPa gel premix, add 1% (1.67 µL) of beads, vortex and sonicate for 5 min in a bath sonicator (standard bench ultrasonic cleaner with power of 50–100 W and frequency 40 kHz). Keep the mix protected from light using aluminum foil.
NOTE: The premix does not polymerize until the initiator (TEMED) is added. - To catalyze polymerization, add 1% (1.67 µL) of 10% w/v ammonium persulfate (APS).
- To initiate polymerization, add 0.1% (0.2 µL) N,N,N′,N′-Tetramethylethylenediamine (TEMED). Mix with a pipette. Once APS and TEMED have been added, the gel rapidly polymerizes so proceed quickly to gel casting.
- Gel casting
- Pipet 9 µL of gel mix onto each coverslip/glass-bottom dish (drop in the center, Figure 1A)
- Place the silanized/hydrophobic coverslip and flatten the gel (Figure 1B). Using forceps, press the coverslip to ensure the gel spreads across the entire area of the coverslip (Figure 1C) until it starts leaking out.
- Invert the coverslip/glass-bottom dish into a large Petri dish and tap it on the bench to force beads going towards the gel surface (Figure 1D).
- Cover with aluminum foil and leave for 1 h to polymerize at room temperature in a humid chamber (i.e., put a wet tissue above the dish to prevent evaporation).
- After 1 h, add PBS to the sample to facilitate coverslip release. Carefully, remove the coverslip using a needle (the coating with different silanes should allow easy peeling off of the coverslip from the gel, Figure 1E).
- Leave the gel in PBS.
NOTE: Gels can now be stored in PBS at 4 °C for 5–7 days, but it is recommended to use them within 48 h.
2. Gel functionalization
- Prepare sulfosuccinimidyl 6-(4'-azido-2'-nitrophenylamino)hexanoate (Sulfo SANPAH) solution at 0.5 mg/mL in 10 mM HEPES buffer. This can be stored at 4 °C covered with aluminum foil for up to one week.
- Aspirate the PBS from gels.
- Add 150 µL of Sulfo SANPAH to the gel at room temperature (Figure 1F).
- Expose the gel to UV treatment for 2 min to photoactivate the sites of Sulfo SANPAH and make it stick to the gel surface.
- Wash with PBS three times (Figure 1G).
- Repeat steps 2.2–2.5.
- Add 250 µL of HEL (100 µg/mL) to each gel and incubate overnight in a humid chamber at 4 °C overnight while keeping covered with aluminum foil (Figure 1H).
- Remove HEL antigen and wash with PBS three times.
NOTE: HEL acts both as an antigen and as an adhesion molecule. It can be replaced by other molecules that bind to the receptor (e.g., an anti-mouse IgM, Bovine Serum Albumin, Ovalbumin) or mixed with integrin ligands (e.g., ICAM1 binding to LFA1). If needed, antigen extraction can be observed with a fluorescent version of the HEL (obtained by staining the molecule with a protein labeling kit, see step 4). Note that a given concentration in bulk might not yield the same surface concentration on the gel as on the glass: this needs to be quantified with secondary staining if direct comparison is required.
3. Cell loading and imaging
- Before imaging, remove PBS from the gels and add 500 µL of B cell media (RPMI 1640, 10% decomplemented fetal calf serum, 1% penicillin-streptomycin, 2% Sodium Pyruvate, 50uM Mercaptoethanol and 1X Non Essential Amino Acids) and let them to equilibrate to RT.
- Cell preparation
- Purify primary B cells from spleen according to a negative selection protocol (see Table of Materials). Typical final B cell yield is around 1 x 107 cells. Concentrate this to 3 x 106 cells/mL in B cell medium (RPMI-1640 supplemented with 10% fetal calf serum, 1% penicillin–streptomycin, 0.1% mercaptoethanol and 2% sodium pyruvate).
- Store cells as needed for up to 6 h at 4 °C.
- Keep the cells at 37 °C for 30 min before image acquisition.
- Imaging
- Use a confocal microscope with thermal and (possibly) CO2 control.
NOTE: Regardless of whether a confocal or spinning-disk microscope is used, it is important to use an objective/pinhole that allows a pixel size <200 nm to comfortably track the beads in the analysis phase (e.g., 60x, NA 1.3). Epifluorescence microscopy can also be used, however it provides lower signal to noise ratio and may make individual bead tracking harder. - Two main layers of beads will appear on the bottom and the top of the gel. Focus on the gel plane.
NOTE: A nice gel will appear as a starry sky, with beads approximately uniformly distributed on the same plane. - Program the acquisition for 30 min with a frame rate of 5 s (this is adaptable to the needs of the experiment, e.g., acquire other colors, acquire z stack, etc.)
- Aspirate the media from the gel, leaving about 200 µL of media on the gel. Position the gel on the microscope and find the surface layer of beads and a nice even area on the gel.
- Add 80 µL of cells (avoid touching the gel to maintain focus).
- Ensure that the focus is still correct and that cells can be seen descending in the area (under transmitted light). Launch the acquisition before the cells reach the gel.
- In case of accidental contact with gel, vibrations, or focus drift, adjust the focus.
NOTE: It is crucial to collect an image of the relaxed gel and this can be any image taken before the arrival of the cells on the gel.
- Use a confocal microscope with thermal and (possibly) CO2 control.
4. Fluorescent HEL extraction experiment
- Prepare fluorescent HEL by binding a fluorescent dye (of a color different from the beads one such as Alexa 555), see the Table of Materials.
- In step 2.7, replace conventional HEL with the fluorescent HEL.
- Acquire images with low illumination settings or low frame rate (e.g., 2 frames per minute) to avoid photo-bleaching.
- To quantify HEL extraction, compute the intensity integrated over the cell area for each frame I(t) corrected and normalized by the intensity I(0) of frame 0 according to the formula:
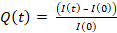
NOTE: The antigen conjugated with a fluorophore is not visible (probably due to quenching of the fluorophore at the gel surface), but its presence on the gel can be verified with an anti-HEL and a fluorescent secondary antibody. It can be verified that the fluorophore is indeed fluorescent when detached by stripping it from the gel with a coverslip coated with anti-HEL and revealing it with a secondary fluorescent antibody (on the coverslip)6. The signal of the extracted antigen is very dim and is sometimes masked by leaking of the beads. If one is interested only in antigen extraction, it is recommended to prepare the gel without beads (skip steps 1.3.2 and 1.4.3).
5. Fluorescence imaging
- Obtain fluorescent B cells by purifying B cell from the spleens of genetically modified mice as done for the wild type (e.g., from Lifeact-GFP or Myosin II GFP mice).
- For imaging fluorescent cells, use (if possible) a spinning disk microscope with a water immersion long-distance 40x–100x objective.
- Keep exposure duration and frame rate low to avoid bleaching.
NOTE: The point spread function in Z is highly degraded by the presence of the gel, hence we suggest using a water immersion objective. Live upright microscopy with water-dipping objectives suffers from strong spherical aberrations induced by the presence of the (spherical) cell (and cell nucleus) in the emission path.
6. Analysis
NOTE: Data analysis is in general performed by first correcting the whole stack for drift, finding the beads in each frame, tracking their movements with respect to a reference frame (taken in absence of cells), interpolating the displacement field and inverting the problem to obtain the stress using Fourier transform29. To this end, we suggest using a combination of ImageJ Macro and MATLAB programs downloadable from an online repository30.
- Open the movie in ImageJ as stack of images
- Run the macro “Crop_and_save.ijm”
- Select the regions of interest (ROI) with the “Rectangle” tool and add them to the ROI list using the ‘t’ key.
- When cropping the cell, be sure to include a region of at least 5–10 pixels of immobile beads. Exclude cells that are too close to the boundaries or to other cells from the analysis. When finished click on ‘OK’.
- The macro proposes a mask of the cell: if this is satisfactory click on “OK”. If not satisfactory, click on “Not ok” and then manually select a closed region with any selection tool (e.g. “Freehand” or “Oval”) and click on “Continue”.
- Open MATLAB and run “TFM_v1.m”.
- Input the required parameters: in particular check the image properties (pixel size, time interval of acquisition) and the gel properties (Young modulus E, Poisson ratio).
- The reference image is set to be the first by default. Set it to another frame if needed or set it to “0” to load an external file.
- Locate the outputs of the software in the same directory as the original file (for a description see the User_notice.pdf file). This includes a preliminary track of the beads (“FILENAME.fig”), a plot of the contractile energy over time (“FILENAME_energy.fig”), a table of several quantities integrated over the cell (energy, area, moments, etc) “FILENAME_finaltable.mat”, a structure containing the displacement and force field, movies of the bead, displacement field, stress and energy (that can be opened with any avi reader).
NOTE: In the input parameters, the “Window size” is the window over which the displacement is interpolated, hence the final resolution of the stress and displacement field. This is set to a few (by default four) pixels. It is not advisable to reduce this as it would artificially increase the resolution by interpolating regions where there are no beads.
Given the size of the cells, algorithms that extract the displacement map of the beads via correlative techniques (such as particle image velocimetry) are in general not very precise. However depending on the degree of resolution required, one can easily obtain qualitative results using a free Fiji/ImageJ plugin31,32. While this approach is sufficient to compare stimulating versus non-stimulating conditions, for a thorough analysis we recommend using our software downloadable from an online repository30, that tracks the beads individually and provides the displacement field map at a given time point as the interpolation of the individual bead displacements33. Several quantifications are possible at this point. For example (by assuming the displacement is caused only by stress tangential to the gel surface) the software also provides the stress at each point causing that specific displacement map. This is a type of “inversion problem”: the displacement at a certain point depends on the sum of all the forces applied all over the other points. The “inversion algorithm” takes into account the physical parameters of the substrate: its rigidity (Young modulus) and Poisson ratio. Direct algorithms are typically very accurate but computationally expensive. Algorithms based on Fourier transform, like ours, perform essentially a deconvolution in Fourier space and are more efficient but prone to some errors (mainly due to the interpolation step). These algorithms generally require the tuning of a parameter that prevents small local (and potentially artifactual) displacements to become too relevant in the computation of the stress field (Tikhonov regularization parameter8,29; “Regularization” variable in the dialog window; here we typically set equal to 5 x 10-19). For more advanced interpretation and analysis, such as spatio-temporal correlations, local movements, correlations with fluorescent channels, we recommend collaborating with experts in the field. For a review on computational methods see Schwarz et al.9.
As mentioned above, correct bead images look like a “starry sky”, a uniform and random distribution of bright spots (Figure 2A). Data and analysis are not reliable when the number of beads is too low (Figure 2B) or the image is out of focus (Figure 2C). Once B cells have settled onto the surface of the gel, the beads underneath the cells start to move due to the traction force exerted by the cell on the gel. Frames for which the beads are not trackable should be discarded.
As a check, it is possible to observe by eye the movement of beads comparing the “reference frame”, typically the one preceding the first contact of the cell with the substrate. Approximate results can be obtained from the single particle tracking (e.g., Trackmate, Fiji 34) as done in Figure 3A. The analysis provides a segmentation of the beads in the reference image (“FILENAME.fig”) as a control.
With the software we propose, one can obtain the displacement (Figure 3B) and stress field (the vector of the local stress at each pixel and each time point obtained by inversion from the displacement field, Figure 3C). Scalar product of the displacement and force fields integrated on the area of the cell provides total work exerted by the cell on the substrate (Figure 4A). This computation requires the mask of the cell introduced in step 6.2 of the protocol.
To compare two biological conditions (as activating HEL versus non-activating substrate BSA, or wild type versus knock-out) it is useful to compute the average curve (Figure 4B) or, even more synthetically, an average value over the last time points (20 min) where the energy reaches a plateau (Figure 4C). When the spatial information of the forces is relevant it is possible to compare single time points of each condition (Figure 4D). Refer to Kumari et al.6 for deeper analysis.
An example of fluorescence antigen extraction time lapse is shown in Figure 5A: the progressive appearance of fluorescence signals at the synapse indicated antigen detachment from the gel. The average extraction curve with its confidence interval (standard error of the mean) over 15 cells is shown in Figure 5B.
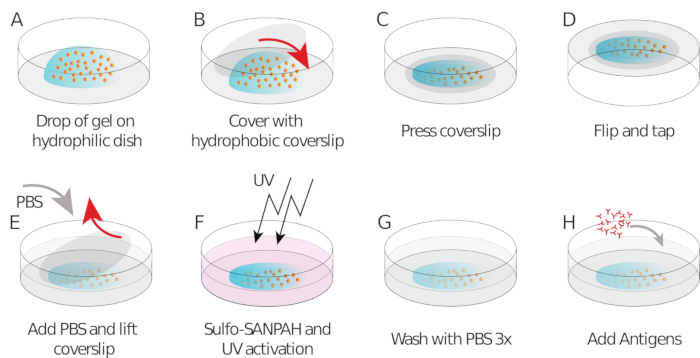
Figure 1: Schematic showing of the preparation of the gel and its functionalization. Steps are described in the protocol. Please click here to view a larger version of this figure.

Figure 2: Three examples of bead images of different qualities. (A) Example of bead image with the correct signal to noise ratio and the correct density. (B) Examples of images with a too insufficient number of beads and (C) out of focus plane. Please click here to view a larger version of this figure.
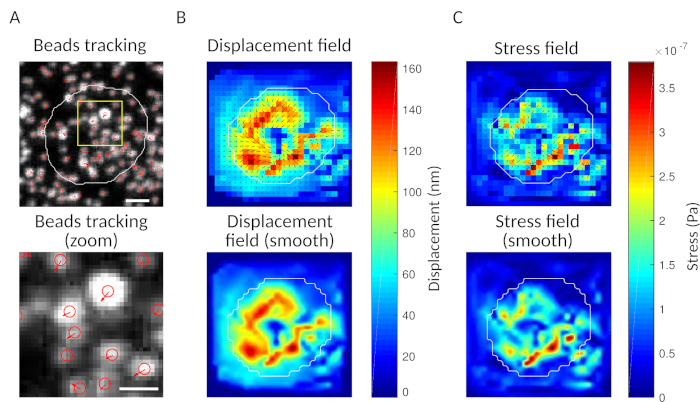
Figure 3: Processing of the images to extract the force field. (A) Example of an image of the beads (outline of the cell in white, extracted from the transmission image), bead tracking at time t = 5 min (red overlay) and displacement (arrows) relative to time t = 0 min (scale bar 5 µm). (B) Interpolated displacement field (represented as vector quiver and magnitude map, arrows are proportional to the displacement [nm]; see the color bar on the right); bottom: a smoother image of the magnitude (obtained by interpolation with a bicubic function). (C) Stress field from displacement field in panel B (represented as vector quiver and magnitude map; arrows are proportional to the shear stress [Pa]; see the color bar on the right); bottom: a smoother image of the magnitude (obtained by interpolation with a bicubic function). Please click here to view a larger version of this figure.
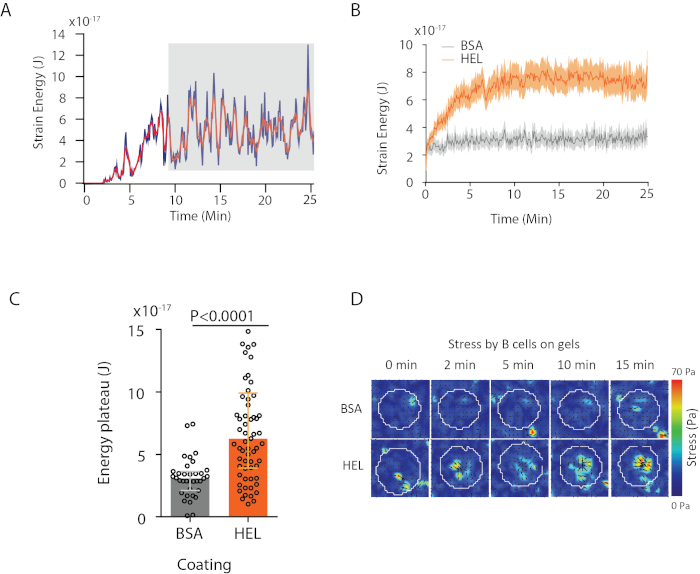
Figure 4: Example of information that can be extracted from force and displacement fields. (A) Example of evolution of energy in time for a single cell: a plateau phase (highlighted in gray) shows up after about 10 min. (B) Comparison of the average energy curves and (C) of the relative plateau levels for 65 cells plated on HEL (activating) coated gel and 35 cells on BSA (non-activating) coated gel (median ± interquartile ranges are shown, Mann-Whitney test was used for statistical significance). (D) Time-lapse color maps of stress for HEL and control BSA condition; both magnitude and quiver plots are shown. These images have been adapted from Kumari et al.6. Please click here to view a larger version of this figure.
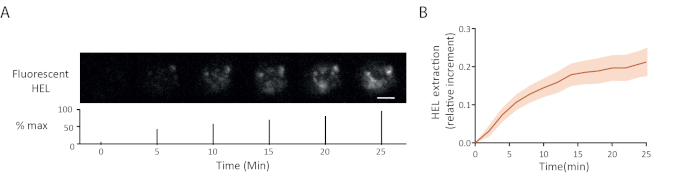
Figure 5: Example of experiments with fluorescent antigen. (A) Time lapse of the extraction of fluorescent HEL (below: percentage of the maximum, scale bar = 3µm). (B) Antigen gathering over time (Mean ± SEM, n = 15). These images have been adapted from Kumari et al.6. Please click here to view a larger version of this figure.
