Driven by the increasing application of next-generation sequencing techniques, spatial resolution in developmental biology could be augmented. Contemporary studies aim at dissecting tissues down to specialized cell types, if not single-cell level1,2,3,4. To this end, a plethora of different methods has been devised over the last fifty years (see Figure 1A)5,6,7,8,9,10,11,12,13,14,15.
Many tools in plant science have been adaptations of techniques that were pioneered in animal research. This is not the case for the method we are introducing in detail here. In 2005, equipped with a strong background in protein translation, the Bailey-Serres Lab set out to engineer ribosomal proteins for subsequent affinity purification16. Thus, they could avoid time-consuming and labor-intensive polysome profiling, which is based on ultracentrifugation with a sucrose gradient and was used to assess translating ribosomes since the 1960s17,18. The method has since been referred to as translational ribosome affinity purification (TRAP)16. After successful translatome studies in plants, Heiman et al. adapted TRAP for animals19 and others extended its application to yeast20, Drosophila21, Xenopus22 and zebrafish23,24.
Although genetic modification of the model system is a prerequisite for TRAP, which limits its application to species amenable to genetic transformation, one can simultaneously harness this objection to target subsets of cells that are of special interest and otherwise extremely difficult to isolate from the intact tissue/organ25 (e.g., highly branched dendritic cells in a mouse brain or fungal hyphae in infected plant tissue). In plants, all cells are held in place via cell walls that form the basis of the hydrostatic skeleton26. To free a plant cell from this matrix, scientists have either physically cut the cell out of its surrounding tissue through laser capture microdissection (LCM)27 or performed enzymatic digestion of the cell walls28. Among the latter cells, so-called protoplasts, the population of interest is fluorescently labeled and can be separated via fluorescence-activated cell sorting (FACS)7. LCM usually requires a sample to be fixed and embedded in wax, which ultimately deteriorates the quality of its RNA29. FACS-based methods yield high-quality RNA, but the process of protoplasting itself introduces differences in gene expression30 and tissues with modified and thick secondary cell walls are notoriously difficult to treat. Moreover, many developmental processes in plants are assumed to rely on mechanically transmitted signals and therefore the integrity of the cell wall is of paramount importance31. Two methods, which use a shortcut to circumvent cell isolation by operating on the level of nucleii, are fluorescence-activated nuclear sorting (FANS) and isolation of nuclei tagged in specific cell types (INTACT). As in TRAP, they use cell type-specific promoters to mark nuclei, that subsequently get enriched via sorting or pull down, respectively8,15. A major challenge for all these approaches is to get sufficient RNA material from subsets of cells in a tissue. As TRAP captures only a fraction of the cellular RNAs, sample collection is a considerable bottleneck. Therefore, especially sensitive library preparation protocols are needed to produce high-quality data from low input amounts.
Since its establishment, TRAP has been either used in combination with DNA microarrays or, as sequencing costs dropped significantly in recent years, RNA-seq10,32,33. A multitude of research questions has already been elucidated as reviewed in Sablok et al.34. We are convinced that more reports will follow in coming years as the technique is very versatile when combining different promoters to target specific cell types. Eventually, this will be done even in an inducible way, and may be combined with probing the plant's reaction to many biotic and abiotic stress factors. Additionally, where stable transgenic lines are not available, hairy root expression systems have also been successfully used to perform TRAP in tomato and medicago35,36.
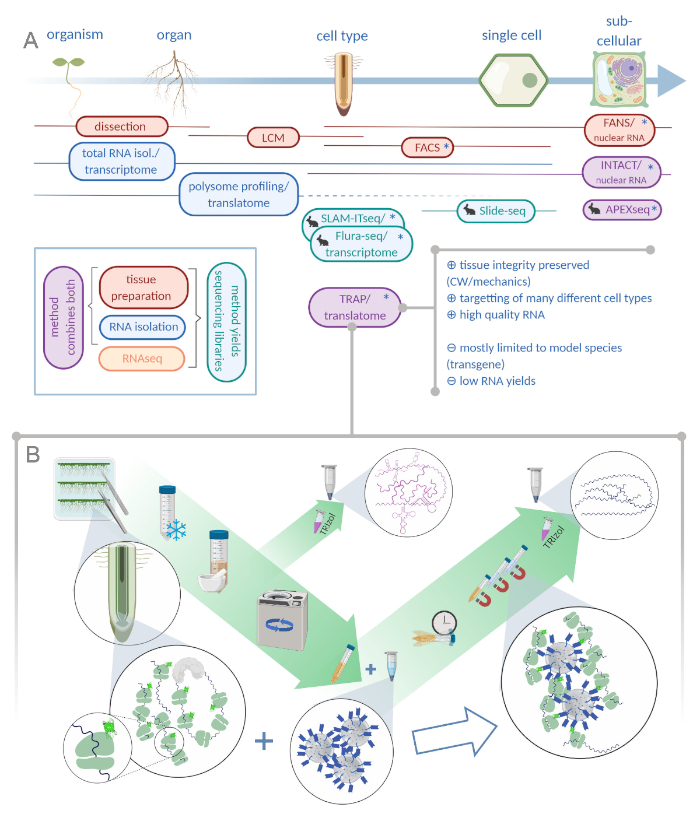
Figure 1: Translating ribosome affinity purification (TRAP) complements the "omics" analysis portfolio. A. Increasing levels of analytical precision, down to single-cell or even subcellular resolution can be achieved by a plethora of methods or combinations thereof. The scheme gives an overview of currently available tools in the plant and animal field. Tissue collection at cellular resolution can be achieved by protocols like LCM or FACS, which are then coupled to standard transcriptome or polysome profiling/translatome analysis. TRAP and INTACT integrate both tissue capture and RNA isolation as they are based on epitope-tagging. However, INTACT samples only cell nuclei and constitutes, therefore, a special case of transcriptome analysis. A small rabbit icon marks newly developed methods in the animal field: While SLAM-ITseq and Flura-seq rely on metabolic targetting of nascent RNAs with modified uracil bases in cells expressing the permissive enzyme, Slide-seq makes use of a coated glass slide with DNA barcodes that provide positional information in the cellular range. A proximity-labeling approach is followed in APEX-seq to sample RNAs in specific subcellular compartments. Notably, increased resolution often requires the generation of transgenic material (asterisks) and these methods are thus predominantly used for model species. TRAP is especially suited for plant science studies involving cell wall (CW) or mechanic signaling as well as cell species that are difficult to release from their CW matrix. B. Detailed wet-lab steps of the TRAP procedure: Seedlings expressing GFP-tagged ribosomal protein in distinct cell types (e.g. root endodermis) are grown on Petri dishes for seven days and root material harvested by snap freezing. A total RNA control sample is collected from the homogenized crude extract before pelleting the debris via centrifugation. Magnetic anti-GFP beads are added to the cleared extract to perform immunoprecipitation. After incubation and three wash steps, the polysome-associated RNA (TRAP/polysome RNA) is directly obtained via phenol-chloroform extraction. LCM: laser capture microdissection, FACS/FANS: fluorescence-activated cell/nuclear sorting, APEX-seq: method based on engineered ascorbate peroxidase, INTACT: isolation of nuclei tagged in specific cell types, SLAM-ITseq: thiol(SH)-linked alkylation for the metabolic sequencing of RNA in tissue, Flura-seq: fluorouracil-labeled RNA sequencing (Created with Biorender.com) Please click here to view a larger version of this figure.
The goal of this article is to supply a detailed description of the TRAP method, to highlight critical steps and to provide guidance for a possible library preparation method.
A generic TRAP experiment will essentially consist of the following steps (see also Figure 1B): (1) Preparation of plant material including cloning of ribosome-tagging construct, transgenic line production and selection, growing and bulking up of seeds, sterilization and plating, and stress application/treatment (optional) and tissue harvesting; (2) immunopurification including tissue homogenization and clearing of the crude extract, bead wash and immunopurification, and wash steps; (3) RNA extraction and quality assessment; and (4) library preparation.
The Arabidopsis root has been a model system to study plant development ever since its introduction as a model plant37,38. Here, the application of TRAP is showcased in the context of plant lateral root development. In plants, the buildup of the entire root system relies on the execution of this program and is therefore very important for the survival of the organism39. In Arabidopsis, lateral roots originate from pericycle tissue that resides next to xylem vessels and therefore is termed xylem pole pericycle (XPP; see Figure 2C)40. Some XPP cells, which are located deep inside the root, acquire a founder cell identity and, upon a local hormonal trigger, start to proliferate by swelling and dividing anticlinally41. However, due to the presence of a rigid cell wall matrix, this process exerts mechanical stress on the surrounding tissues. In particular, the overlying endodermis is affected, as it is in the way of the lateral root growth axis42,43,44. Indeed, the newly forming primordium will have to grow through the overlying endodermis cell (Figure 2C2) whereas cortex and epidermis cells are just pushed aside for the primordium to finally emerge45,46. Recent work in our lab has shown that the endodermis is actively contributing to accommodate the proliferation in the pericycle. Targeted blocking of endodermal hormonal signaling is sufficient to inhibit even the very first division in the XPP cells47. Hence, pericycle-endodermis communication constitutes a very early checkpoint for lateral root development in Arabidopsis. It is, however, not known how this crosstalk is performed. To unravel this mystery, we chose the TRAP-seq approach to target XPP and endodermal cells. To enrich for cells in the lateral root program, we mimicked the hormonal trigger by exogenously applying an auxin analog (1-naphthaleneacetic acid, NAA)48, which at the same time allowed to temporally resolve the initial phase of lateral root formation.
For quality assessment, the above-mentioned procedure should be probed at several intermediate steps: expression pattern validation in planta, quality control of the isolated polysomal RNA as well as of the final libraries. qRT-PCR using known marker genes can, in addition, be performed to confirm the response to the treatment condition or to fine-tune the experimental conditions.
Confocal analysis of GFP signal distribution
To check for both endodermal and XPP expression patterns, we analyzed homozygous lines of pELTP::GFP-RPL18 and pXPP::GFP-RPL18 by confocal microscopy. Figure 2A and Figure 2B show representative plants with GFP signals (green) that have been counterstained with propidium iodide (magenta) to outline cell walls. The cross-section in Figure 2A1 shows a concentric ring in the third cell layer from the outside, which corresponds to the endodermis. The endodermal GFP signal initiates shortly above the meristematic zone (Figure 2A2) and appears both in the cytosol and around the nuclei of the cells, which corresponds to ribosomes. In contrast, the XPP line exhibits two distinct poles, which corresponds to the XPP (Figure 2B1). Approximately three cells at each pole start above the meristematic zone to exhibit a GFP signal. Thus, both lines comply with the localization pattern of endodermis and XPP, respectively (Figure 2C)53.
Polysome RNA validation
To determine the quality of the obtained polysome RNA we performed quality control measurements, using two automated electrophoresis systems (Table of Materials) that work with µL input amounts and also calculate an RNA integrity number (RIN)54. The proprietary algorithm assigns a RIN value between 1 and 10 to each electropherogram and is a robust and reproducible measure for RNA quality (i.e., degradation) – the lower the value the more degraded is the sample. Figure 3 shows examples of the measurements we obtained from polysome RNA. Most samples show hardly any apparent degradation with RIN values ranging from 9-10, which is in accordance with previous reports55,56. Any improper handling, especially periods of prolonged elevated temperatures (e.g., room temperature) or RNase contamination would be evident at this stage. Both instruments also calculate sample concentrations from their electropherograms (Figure 3A). These can vary substantially and are mostly at the lower detection limit. We, therefore, advise using fluorometric measurements to accurately quantify concentrations.
Library QC
As most labs do not perform the RNA sequencing in house, quality controls are often run at specialized facilities with high throughput devices (Table of Materials). They routinely assess the quality and quantify concentrations by qPCR and fluorometric assays (Table of Materials) as accurate measurements are prerequisite for library pooling. Nevertheless, if library preparation is not outsourced, one can sample the outcome with specialized equipment (Table of Materials). Figure 4 shows traces of successfully prepared libraries with our recommended protocol (A) and highlights the robustness of the procedure despite scaled-down reaction volumes (B). Part C illustrates sub-standard samples that can result from over-/underfragmentation, material loss during clean up or unsuccessful adapter removal. In the latter case, another clean up with a more stringent sample-bead-ratio could help eliminate the contamination. Completely failed samples were extremely rare in our hands and could originate at multiple points (e.g., too high input for the stochiometric tagmentation reaction).
The performance of sequenced libraries is exemplified in Figure 4D for samples from the endodermis in our mutant background. Libraries from WT and/or pericycle perform similar or even better. Ribosomal reads were on average around 2% with only few samples above 3%. Reads with an average quality score above 30 were consistently above 90% already before the filtering. The mapping ability of the sequenced reads was equally high ranging on average at 85%. To determine the correlation between biological replicates pairwise Spearman coefficients were calculated for each time point. All tests resulted in high coefficient values.
Treatment response and enrichment analysis
Before a genome-spanning dataset is produced, TRAP RNA from a pilot experiment can be probed by qRT-PCR to validate treatment success and/or experimental conditions. We performed this type of analysis to assess auxin responses after 2 h of treatment in the XPP samples (Figure 5A). Three different auxin-responsive genes (GH3.3, LBD29 and GATA23) were tested via the ∆∆Ct method57. Very strong induction was observed in all three cases after the incubation period, which suggests that the exogenous NAA application was successful.
If newly developed promoters are utilized one should at this point also perform enrichment analysis with qRT-PCR. To this end, a known marker gene (i.e., the gene driven by the promoter used) is amplified in the TRAP and total RNA sample and expression levels normalized to the total RNA level. If the isolation of TRAP RNA from the specific tissue was successful a significant fold change increase should be obtained. Alternatively, equivalent information can be retrieved from the sequencing data (see Figure 5B). Expression of two suberin-related genes, GPAT5 and HORST, is present in all endodermis samples and notably absent from the XPP tissues. On the contrary, pericycle-expressed genes (PHO1 and SKOR) are only very lowly expressed in the endodermis and enriched in the XPP probes with an auxin-induced down-regulation over the examined time frame.
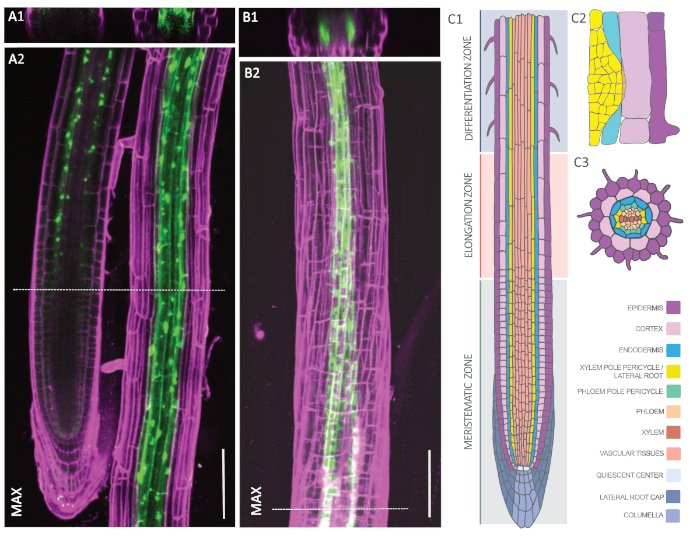
Figure 2: Cell type-specific expression of GFP-RPL18 in the Arabidopsis root. A-B. Confocal microscopy images of pELTP::GFP-RPL18 (A) and pXPP::GFP-RPL18 (B) expressing roots at six days post germination. Cell wall outlines were obtained through staining with propidium iodide (magenta). Cross-sections A1 and B1 are from the positions denoted with dashed lines in A2 and B2, respectively. The latter images show maximum projections (MAX) of the recorded z-stacks. C. Schematic representation of the tissue types composing the Arabidopsis root in longitudinal (C1) and cross-section (C3) as well as in a lateral root primordium (C2). The image was modified with permission from F. Bouché. Scale bars: 100 µm. Please click here to view a larger version of this figure.
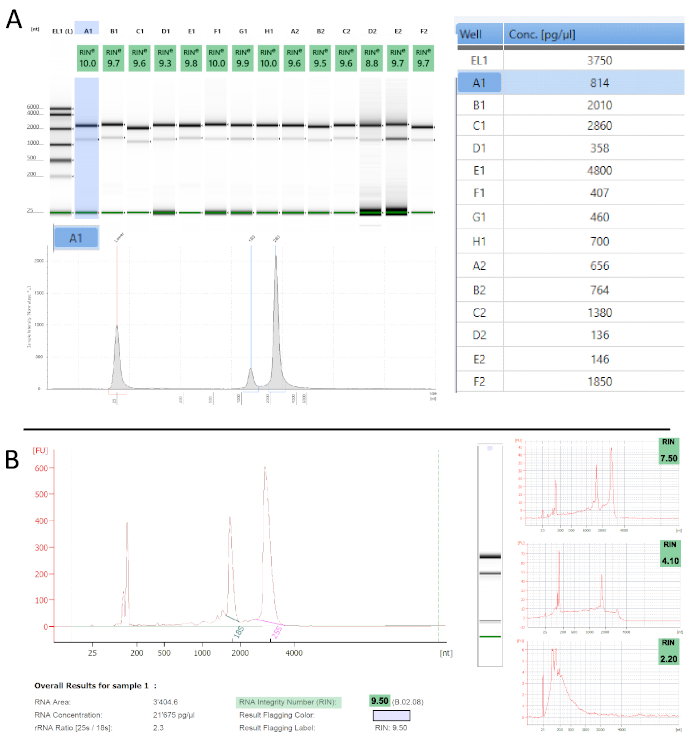
Figure 3: TRAP/polysome RNA quality assessment. A. Tapestation – Representative results from 14 measured samples in gel picture representation with their respective RINe values (top left). Electropherogram representation is shown for sample A1 (highlighted in blue). The table on the right informs about the sample concentrations. B. Similar traces as in A are obtained with the Bioanalyzer. The panels on the right show samples with increasing levels of degradation, which reflects in their decreasing RIN values. Please click here to view a larger version of this figure.
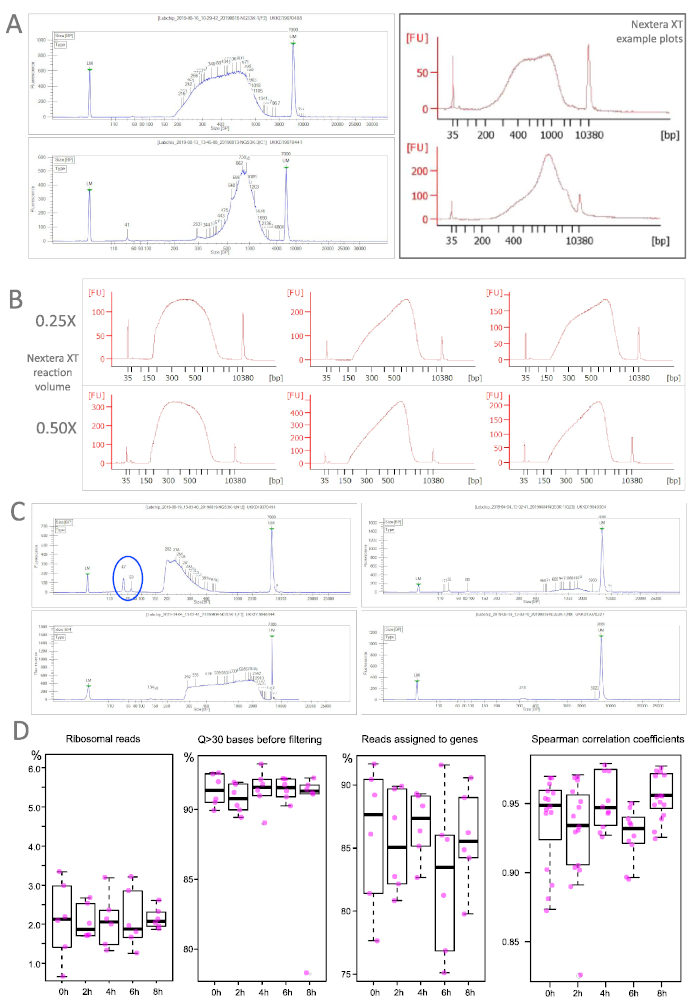
Figure 4: Library profiles from TRAP/polysome samples. A. Two representative TRAP samples (left) correspond very well with the traces for successful libraries recommended by the Nextera XT user guide. B. Differing reaction volumes yield robust library preparation outcomes. C. Libraries with suboptimal outcomes: very short fragments (top left), extremely long fragments (bottom left), low concentration (top right) or complete fail (bottom right). Note also residual short fragments (blue ellipse), that have to be removed before sequencing. Bioanalyzer: red traces, LabChip: blue traces. D. Selected quality measures for sequenced TRAP samples (endodermis of our lateral root-free mutant) at different time points and distribution of Spearman correlation coefficients calculated between pairwise comparisons of all samples within a time point (n=65). Please click here to view a larger version of this figure.
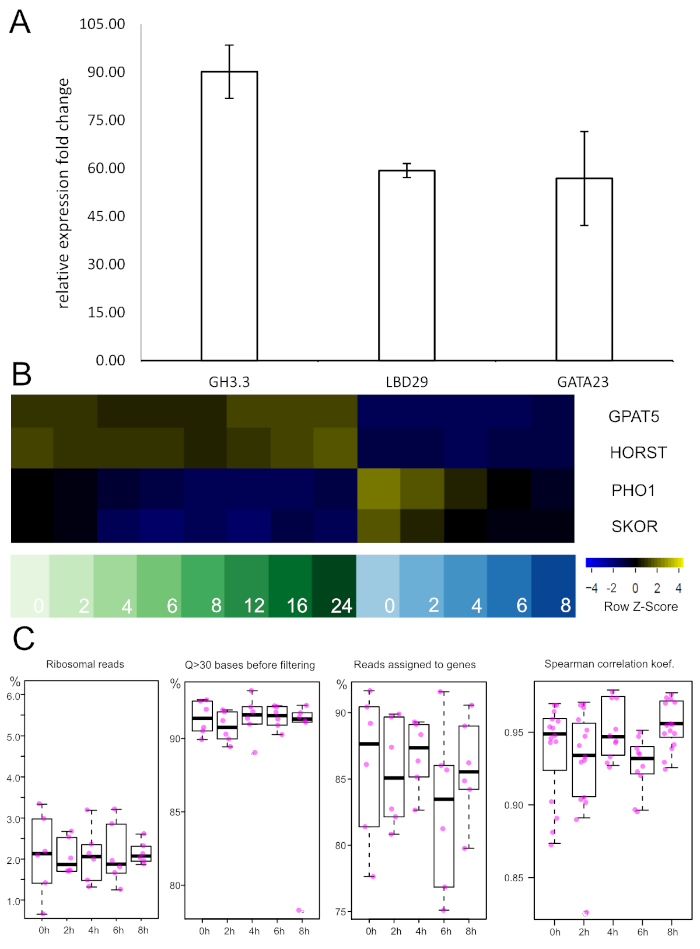
Figure 5: qRT-PCR and RNA-seq show auxin-responsiveness and tissue type enrichment, respectively. A. Expression levels of three known auxin-responsive genes were assessed after 2 h of auxin treatment via qRT-PCR. Strong induction was observed in all samples. RT-PCR was performed on 3 independent biological replicates and normalized to the non-treated samples with UBC21 as internal reference gene. Error bars represent the SEM. GH3.3: Gretchen Hagen 3.3, LDB29: LATERAL BOUNDARY DOMAIN 29, GATA23: GATA-motif binding transcription factor 23, UBC21: UBIQUITIN-CONJUGATING ENZYME 21 B. Expression levels of four marker genes from the TRAP-seq dataset. Samples on the left are endodermis-derived (green shades), while samples on the right are XPP-derived (blue shades). Numbers represent the auxin incubation intervals in hours. Negative z-scores reflect low expression levels and vice versa. Endodermal marker genes (GPAT5, HORST) are differentially expressed with high levels in endodermis samples. On the contrary, pericycle markers (PHO1, SKOR) have high expression levels in XPP cells and show down-regulation upon auxin treatment. GPAT5: glycerol-3-phosphate 2-O-acyltransferas (suberin biosynthsis), HORST: hydroxylase of root suberized tissue, PHO1: phosphate 1, SKOR: stelar K+ outward rectifier. Please click here to view a larger version of this figure.
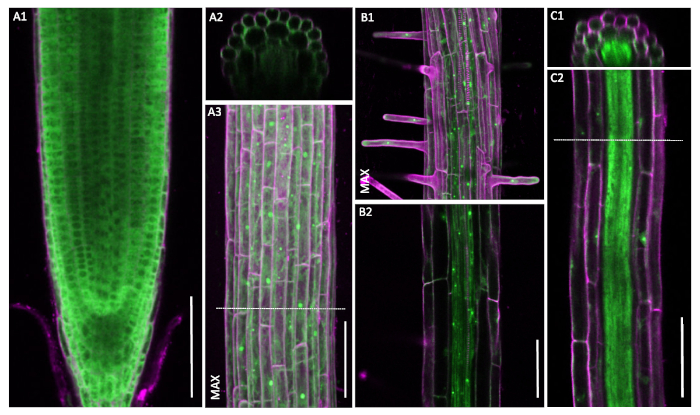
Figure 6: Non-constitutive pUBQ10::GFP-RPL18 localization patterns. Confocal microscopy of six-day-old seedlings. Cell wall outlines were obtained through staining with propidium iodide (magenta). Cross-sections A2 and C1 are from the positions denoted with dashed lines in A3 and C2, respectively. Images marked MAX show maximum projections of the recorded z-stacks. A1-A3. Uniform localization patterns of the UBQ10-driven construct. B1-C2. Notable decrease in the signal strength in outer tissue layers. A, B and C are recorded in three different plants. Scale bars: 100 µm. Please click here to view a larger version of this figure.
