All methods have been approved by the Institutional Animal Care and Use Committee (IACUC) of East Carolina University.
1. Ozone (O3) and filtered air exposures (Day 1)
- Place a maximum of 12 female C57BL/6J mice, 8-12 weeks old, in a steel cage (with 12 separate compartments) with wire mesh lids into an O3 exposure chamber.
- Place the thermometer in the exposure chamber with the cage to accurately record the temperature and humidity.
- Turn on the oxygen and ultraviolet (UV) light that is attached to the apparatus.
NOTE: Regulated airflow (>30 air changes/h) with controlled temperature (22-23 °C) and relative humidity (45%-50%) is obtained by the O3 apparatus. O3 is generated by the system in the exposure chamber by directing 100% oxygen through a UV light generator, then mixing with a filtered air supply. - Adjust the O3 concentration to 1 ppm and regularly record O3 levels every 10 min for 3 h. Continuously monitor the temperature and humidity of chamber air, as is the O3 concentration with a UV light photometer.
NOTE: Filtered air exposures are performed in a similar apparatus, with only a filtered air supply flowing through the exposure chamber. - Return the animals to their respective cages with bedding, food, and water ad libitum after 3 h of O3/filtered air exposure.
2. Preparation of Jurkat T cell line (Day 2)
NOTE: All procedures should be conducted in a class II biological safety cabinet.
- Culture Jurkat T cells in 24 mL of basal cell culture medium + 10% FBS + 5% penicillin/streptomycin at 37 °C + 5% CO2 (Table of Material). Jurkat T cells are a suspension cell line that can be maintained through passaging 1:6-1:8 into pre-warmed culturing media every 3 days. Do not shake.
- To prepare apoptotic cells, grow them to 90% confluency in each flask (which takes 3-4 days to achieve after passaging). For this study, use cells from five T75 flasks to obtain the sufficient number of cells used in this protocol.
NOTE: A confluent flask contains about 20-24 million cells. - Pipette up cells (which is the entire flask) from each flask (approximately 24 mL) and transfer cells to a sterile 50 mL conical tube using a serological pipette. Use multiple conical tubes for multiple flasks.
- Count cells by removing an 11 µL aliquot of cells from the 50 mL conical tube and mix with 11 µL of trypan blue stain and pipette 11 µL onto hemocytometer slides.
- Insert slide into an automated cell counter and record the number of live cells to calculate the total cell count in each flask by multiplying the number of live cells by 24, as each flask contains 24 mL of media.
- Centrifuge the cell suspension at 271 x g for 5 min at room temperature (RT) to pellet cells.
- Discard the supernatant by aspiration and resuspend the cell pellet in media to obtain 3.0 x 106 cells per mL.
- Aliquot 5 mL of cells in 100 mm x 20 mm tissue culture dishes (approximately nine dishes will be used; the total amount of cells in each dish should be ~15 x 106).
- Use one dish for control/unexposed, and the remaining dishes will be exposed to UV.
- Set the UV crosslinker to the correct energy level, press the energy button, and enter "600" using the number pad, which the machine will read as 600 µJ/cm2 x 100.
NOTE: UV crosslinker energy units is in µJ/cm2 x 100; therefore, to achieve 60 millijoules/cm2, convert units to match the UV crosslinker. - Irradiate all dishes with cells, not including the control, at 60 millijoules (mJ)/cm2 using the UV crosslinker. Remove the top cover of the tissue culture dishes during UV exposure, as UV light will not penetrate the plastic cover.
- Incubate all the dishes in a cell culture incubator, including unexposed control, at 37 °C at 5% CO2 for 4 h.
- Confirm apoptosis by flow cytometry using an apoptosis assay detection kit containing annexin V and propidium iodide (PI) (markers for apoptosis and necrosis, respectively) after 4 h of incubation, per the manufacturer's instructions26,27.
NOTE: Irradiating Jurkat T cells in the UV crosslinker at an energy level of 600 µJ/cm2, following a 4 h incubation will lead to ≥75% of apoptotic (both early and late) cells having more early apoptotic phenotype than the late apoptotic phenotype. This makes it easier for alveolar macrophages to recognize them and engulf as their membranes are uncompromised, unlike late apoptotic cells, leading to a higher efferocytic index and more accurate imaging of alveolar macrophage efferocytosis in this study.- Pool 333 µL (1 x 106 cells) of Jurkat T cells from several dishes (both "no UV" and "UV-exposed") together to use for compensation analysis tubes.
- Aliquot 333 µL of Jurkat T cells in an unstained, annexin V single stain, PI single stain, no UV control, and 600 µJ/cm2 UV-exposed labeled flow cytometry tubes.
- Centrifuge tubes at 188 x g for 5 min at RT and decant the supernatant.
- Wash cells by resuspending in 500 µL of cold, 1x phosphate-buffered saline (PBS).
- Centrifuge and pellet cells at 188 x g for 5 min at RT. Discard the supernatant after centrifugation.
- Prepare 400 µL of 1x binding buffer per flow tube by diluting 10x binding buffer with distilled water while cells are centrifuging.
- Prepare annexin V and PI incubation reagent (100 µL per sample/tube) per the manufacturer's instructions.
- Decant the supernatant after centrifugation and gently resuspend all tubes in 400 µL of 1x binding buffer, then add 100 µL of annexin V incubation reagent to each sample tube. Lastly, add 100 µL of annexin V single stain and PI single stain to their respective tubes, but do not add anything beyond the 1x binding buffer to the unstained tube.
- Incubate tubes in the dark for 15 min at RT.
- Centrifuge all cells at 188 x g for 5 min at RT and decant supernatant.
- Resuspend cells in 400 µL of 1x binding buffer, then analyze samples for apoptosis by flow cytometry. Collect at least 10,000 events per tube to allow accurate representation of staining.
- Combine all the irradiated cells from dishes into a 50 mL conical tube and pellet cells by centrifugation at 271 x g for 5 min at RT.
- Discard the supernatant from the tube by aspiration and resuspend cells in 24 mL of sterile phosphate buffered saline (PBS) and pellet cells by centrifugation at 271 x g for 5 min at room temperature.
- Discard the supernatant from the tube by aspiration and resuspend cells in the amount of PBS used for dosing mice approved by IACUC. The dose used is between 5-10 x 106 cells/50 µL per mouse; therefore, for 10 mice, resuspend in 500 µL (number of cells in each dose varies depending on how many cells are cultured for irradiation).
NOTE: Makeup at least two additional doses to account for any liquid that may stick to the sides of the pipette tip resulting in the loss of cells.
3. Murine oropharyngeal instillation of apoptotic cells (Day 2)
- Prepare dosing inoculum of apoptotic cells using a P200 pipette prior to anesthetizing mice to expedite the procedure. As per the institutional guidelines, a volume of 50 µL containing approximately 5-10 x 106 cells is utilized for oropharyngeal (o.p.) instillation to ensure best results.
- Anesthetize mice in a clear chamber with 2% isoflurane at a flow rate of 1 L/min or as per the institutional guidelines. Anesthetize one to two mice at a time; the number is determined by the comfort level of the experimenter. Observe the breathing pattern and confirm deep breaths are visible with 2–3 s counts between breaths. Check for the depth of anesthesia by the lack of response to the toe pinch.
- Position the mouse in a semi-recumbent supine position. Use a surgical string tied between pegs on a slanted acrylic sheet board to suspend by the maxillary incisors.
- Using a pair of blunt non-ridged forceps, lightly grab and pull the mouse tongue. Instill the apoptotic cells into the oral cavity with a P200 pipette. Dosing is successful when the mice make a crackling noise 1–2 s after giving the dose.
NOTE: Take care to avoid inducing trauma either to the tongue or oropharynx before the apoptotic cell instillation. - With a gloved finger, gently block the nose until the mouse inhales while the tongue is retracted. Cover the nose until no liquid is visible in the oral cavity and the mouse has taken two or more inhalations.
NOTE: As mice are obligate nose breathers, covering the nose helps ensure that the mouse will inhale the apoptotic cells into the lungs. - Remove the mouse from the inoculation board and return it to the cage to allow recovery from anesthesia. Place the mouse on its back to prevent bedding or debris from blocking the nares during the revovery.
- Wait 90 min after the mouse recovers from anesthesia to allow alveolar macrophages to engulf influx of apoptotic cells after all the mice have awoken from anesthesia. Typically, awakening after anesthesia will take 1–2 min, which should not affect the outcome/timing of instillation.
4. Bronchoalveolar lavage fluid collection and processing (Day 2)
- Euthanize each mouse per institutional guidelines 90 min after the mouse has woken up from anesthesia from dosing with apoptotic cells. Here, a lethal injection of ketamine and xylazine is used (90 mg/kg and 10 mg/kg, respectively) followed by excising the diaphragm.
NOTE: This time point allows sufficient time for alveolar macrophages to sense and engulf apoptotic cells38. - Weigh all mice (g) on a scale and record weights. Use the body weight to calculate BAL volume (26.25 mL/kg body weight).
- Place mice on their backs and spray 70% ethanol to sterilize the chest and neck area.
- Make a 2” longitudinal cut just below the sternum along the entire ventral side with surgical scissors, and while holding the sternum with forceps, nick the diaphragm to allow the lungs to fall back into the chest cavity.
- Cut laterally along the sides of the rib cage to allow the lungs more room to expand when lavaging, then fold the chest cavity back with forceps.
- Make a 1" vertical cut up along vasculature through the neck to expose the trachea.
- Use two forceps to pull muscle and tissue off the trachea and expose it. Avoid additional potential bleeding and cutting the trachea, since it is surrounded by vasculature, longitudinal muscles, and connective tissue.
- Use a needle to make a slit in the trachea (about one-quarter of the distance down from the head) and insert a cannula (18 G x 1.25") with a syringe pre-loaded with 1x PBS (26.25 mL/kg body weight, ~0.7-1.0 mL in an 8-10 week old female C57Bl/6J mouse) caudally into the trachea.
- Push volume of PBS into the lungs slowly to allow the lungs to inflate then, pull the volume back out into the syringe. Repeat this process a total of 3 times.
- Collect the pooled lavage fluid from each specific mouse in a 15 mL tube.
- Centrifuge the bronchoalveolar lavage at 610 x g for 6 min at 4 °C and collect supernatant into a 1.5 mL tube and freeze at -80 °C. The pellet represents cells from the bronchoalveolar space.
- Remove residual red blood cells in collected BAL fluid by adding 1 mL of ACK RBC lysis buffer to the cell pellet, then vortex well and lyse for 1 min on ice. Afterwards, add 4 mL of PBS to stop the lysis reaction.
- Pellet cells by centrifugation at 610 x g for 6 min at 4 °C and aspirate the supernatant with a vacuum aspirator.
- Resuspend cells in 1 mL of 1x PBS + 10% FBS to each BAL sample tube. Count cells on a hemocytometer for the quantification of total airspace cells from each sample (no trypan blue). Centrifuge 120 µL of each sample onto slides at 56 x g for 3 min, using medium acceleration and a cytocentrifuge. Dry the slides overnight.
5. Calculation of alveolar macrophage efferocytic index (Day 3)
- Stain the slides with hematoxylin and eosin to allow for calculation of both efferocytic and differential cell counts, with at least 200 cells counted from each slide.
- View slides under a bright-field setting on a biological microscope (a 20x or 40x objective will work best).
- Calculate the efferocytic index based on the ratio of the number of alveolar macrophages that phagocytosed apoptotic Jurkat T cells to alveolar macrophages without apoptotic cell uptake out of a total 200 macrophages on a cell differential slide. Convert the ratio to a percentage for data input. Use the following equation:
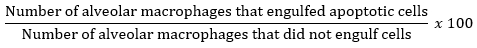
O3 exposure is known to induce pulmonary inflammation and injury, and efferocytosis is required to maintain tissue homeostasis. C57BL/6J female mice were exposed to filtered air (FA) or 1 ppm O3 for 3 h and necropsied 24 h post-exposure to examine pulmonary inflammation and injury. O3-exposed mice displayed a significant increase in macrophages and neutrophils in the airspace compared to the FA control group (Figure 1A,B). Additionally, O3-exposed mice had a significant increase in BAL protein, a marker of alveolar epithelial barrier dysfunction 24 h post-exposure (Figure 1C).
To determine if O3-induced pulmonary inflammation is associated with defects in alveolar macrophage efferocytosis in vivo, C57BL/6J female mice were instilled with apoptotic Jurkat T cells via oropharyngeal aspiration 24 h post-FA or post-O3 exposure. Apoptosis in Jurkat T cells was confirmed by flow cytometry prior to dosing, and there was a significant increase in early (annexin V+ PI– and late (annexin V+ and PI+) apoptotic cells (Figure 2A,B). The exposure level and incubation time resulted in repetitive results of ~75% apoptotic Jurkat T cells. A magnified image of what was identified as an efferocytic macrophage is shown in Figure 3A. Efferocytic macrophages were identified as macrophages that had engulfed a Jurkat T cell (indicated by black arrows), compared to regular alveolar macrophages (indicated by white arrows) (Figure 3B). When alveolar macrophage efferocytosis was assessed utilizing the protocol, there was a statistically significant decrease in the efferocytic index of the O3-exposed group compared to FA controls (Figure 3B,C). These data indicate that O3-induced pulmonary inflammation is associated with decreased clearance of apoptotic cells, which may prolong lung injury and inflammation.
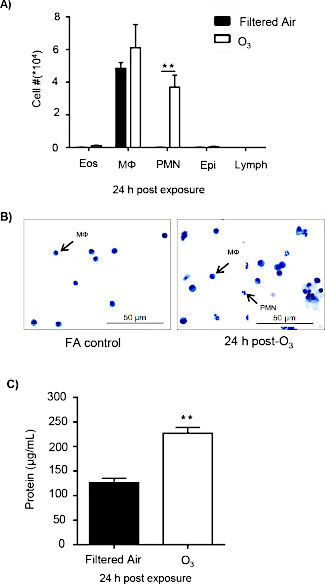
Figure 1: O3 exposure induces pulmonary inflammation and injury. C57BL/6J female mice were exposed to filtered air (FA) or 1 ppm O3 for 3 h. 24 h post-exposure, mice were necropsied to analyze pulmonary inflammation and injury (n = 6 per group). (A) Bronchoalveolar lavage (BAL) cell differentials were calculated, then epithelial (epi), eosinophils (eos), lymphocytes (lymph), macrophages (Mɸ), and neutrophils (PMN) were identified with at least 200 cells counted from each slide. (B) A representative image of cellular differentials. (C) Total protein in the BAL fluid. Data are expressed as ± SEM (**p < 0.01). Please click here to view a larger version of this figure.
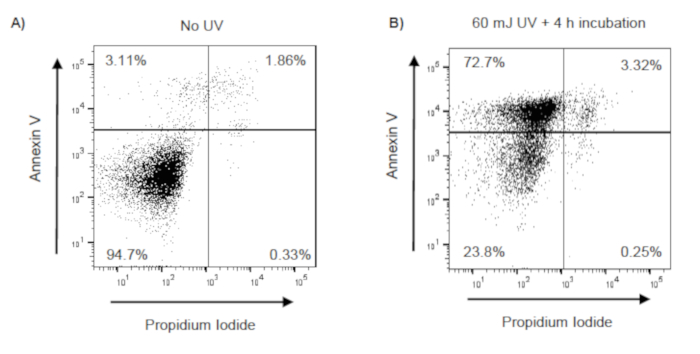
Figure 2: Confirmation of UV induced apoptosis in Jurkat T cells. Jurkat T cells were exposed to UV (60 mJ/cm2) using a UV Crosslinker (Model 1800). Following UV exposure, Jurkat T cells were incubated at 37 °C with 5% CO2 for 4 h. Following incubation, Jurkat T cells were stained with annexin V and propidium iodide (PI), and apoptosis was evaluated by flow cytometry. Early apoptotic, late apoptotic, and necrotic cells are identified as annexin V+/PI–, annexin V+/PI+, annexin V–/PI+, respectively. Representative flow cytometry scatter plots (with 10,000 events recorded) of (A) unexposed Jurkat T cells and (B) UV-exposed Jurkat T cells. Please click here to view a larger version of this figure.
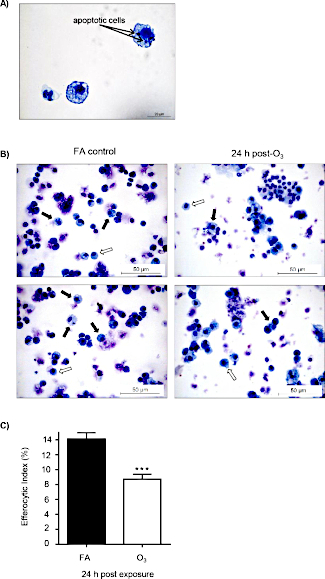
Figure 3: O3 exposure decreases alveolar macrophage efferocytosis. C57BL/6J female mice were exposed to filtered air (FA) or 1 ppm O3 for 3 h. 24 h post-exposure, mice were oropharyngeally instilled with approximately 5 x 106 apoptotic Jurkat T cells. 1.5 h after instillation, bronchoalveolar lavage (BAL) was performed, and the efferocytic index was calculated in BAL macrophages by light microscopy after counting 200 macrophages (n = 11 per group). (A) Representative image of an efferocytic macrophage. (B) Identification of alveolar macrophages (white arrows) and efferocytic macrophage (black arrows) after FA or O3 exposure. (C) Calculation of the efferocytic index after FA or O3 exposure (***p < 0.0001). Please click here to view a larger version of this figure.
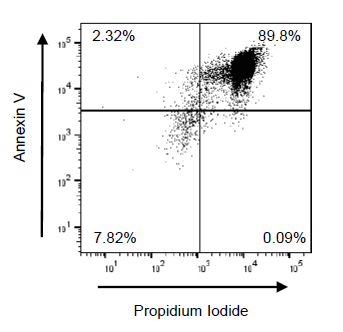
Figure 4: Suboptimal Jurkat T cell apoptosis using 350 nm frosted bulbs. Jurkat T cells were irradiated using the UV Crosslinker for 10 min and incubated at 37 °C at 5% CO2 for 1 h. Following UV exposure, Jurkat T cells were incubated at 37 °C with 5% CO2 for 4 h. Following incubation, Jurkat T cells were stained with annexin V and propidium iodide (PI), then apoptosis was evaluated by flow cytometry. Early apoptotic, late apoptotic, and necrotic cells are identified as annexin V+/PI–, annexin V+/PI+, and annexin V–/PI+, respectively. Representative flow cytometry plots (with 10,000 events recorded) of UV-exposed Jurkat T cells with 350 nm bulbs are shown. Please click here to view a larger version of this figure.
