Mass Isolation and In Vitro Cultivation of Intramolluscan Stages of the Human Blood Fluke Schistosoma Mansoni
Summary
This article describes a protocol for the large-scale axenic isolation and collection of free-swimming miracidia of the human blood fluke Schistosoma mansoni and their subsequent processing for introduction into in vitro culture.
Abstract
Human blood flukes, Schistosoma spp., have a complex life cycle that involves asexual and sexual developmental phases within a snail intermediate and mammalian final host, respectively. The ability to isolate and sustain the different life cycle stages under in vitro culture conditions has greatly facilitated investigations of the cellular, biochemical and molecular mechanisms regulating parasite growth, development and host interactions. Transmission of schistosomiasis requires asexual reproduction and development of multiple larval stages within the snail host; from the infective miracidium, through primary and secondary sporocysts, to the final cercarial stage that is infective to humans. In this paper we present a step-by-step protocol for mass hatching and isolation of Schistosoma mansoni miracidia from eggs obtained from livers of infected mice, and their subsequent introduction into in vitro culture. It is anticipated that the detailed protocol will encourage new researchers to engage in and broaden this important field of schistosome research.
Introduction
The human blood flukes Schistosoma spp., are the causative agents of schistosomiasis, a neglected tropical disease afflicting an estimated 230 million people worldwide1. The most widespread species, Schistosoma mansoni, is geographically distributed in South America, the Caribbean archipelago, the Middle East and sub-Saharan Africa2. The life cycle of S. mansoni, and other schistosomes, is complex, involving a mammalian definitive host and freshwater snails of the genus Biomphalaria that serve as obligate intermediate hosts.
S. mansoni male and female adult worm pairs living in the posterior mesenteric veins of the mammalian host reproduce, resulting in the release of embryonated eggs (ova) that become lodged in the small venules of the intestinal mucosa. Eggs then breaks through the vessel walls, migrate through mucosal tissue, and eventually enter the intestinal lumen where they are voided with the feces. When ova enter freshwater and free-swimming larvae (miracidia) hatch, they must, in short order, find a suitable Biomphalaria snail to infect in order to continue its life cycle. This infection process involves miracidial attachment to the snail's outer body surface followed by active penetration and entry of the larva into the host. Soon after entry, the miracidium begins to shed its outer ciliated epidermal plates as it forms a syncytium that will become the new outer surface (tegument) of the next larval stage, the primary or mother sporocyst. Through asexual reproduction, each primary sporocyst produces and releases a second generation of sporocysts, termed secondary or daughter sporocysts, which in turn, generate and release large numbers of the final intramolluscan stage, the cercaria3. Upon escape from the snail host, free-swimming cercariae are capable of attaching to and directly penetrating the skin of a human or other suitable mammalian host. Upon entry into their new host, cercariae transform to the parasitic schistosomula stage and invade the vascular system. They, in turn, migrate to the lung and then to the hepatic vein where they mature to adult worms, form male and female pairs and travel to the mesenteric veins to complete their life cycle.
Successful intramolluscan or "snail phase" development of larval schistosomes is essential to continuation of the cycle of human host-to-host transmission. Of critical importance is the period following entry of the free-swimming miracidium into the snail host and its early transformation to the primary sporocyst stage. Depending on the physiological and immunological compatibility between host and parasite, it is during this phase of larval development that initial success or failure to establish infections is determined4,5,6. Subsequent development of primary sporocysts capable of asexually producing secondary sporocysts, which then generate human-infective cercariae, further require a permissive physiological host environment that provides for all of the parasite's growth and reproductive needs.
To date, little is known about the underlying physiological, biochemical and molecular processes controlling schistosome-snail interactions, especially the molecules and pathways involved in regulating larval development and reproduction, or modulating host immune responses. Ready access to large numbers of these larval stages under in vitro culture conditions that permit experimental manipulation would greatly facilitate the discovery of developmental pathways and underlying mechanisms of cell growth, differentiation and reproduction. Critical parasite pathways or mechanisms, identified through in vitro experimentation, could then be used to disrupt larval growth/reproduction in the snail host, or to identify snail host immune mechanisms involved in recognition and elimination of miracidial or sporocyst stages.
In this article and video presentation, we provide a detailed description of a method for isolating large numbers of free-swimming S. mansoni miracidia for introduction into in vitro culture that may then be subjected to follow-up experimental manipulation (see Figure 1 for a schematic overview of the protocol workflow). Although similar procedures have been described previously7,8,9, we felt that a detailed protocol would be useful to researchers who wish to employ this model to address still unanswered questions related to intramolluscan larval development, cellular proliferation, and schistosome-snail immune interactions. In addition, this approach could easily be adapted for isolation of eggs from other medically important trematodes such as bladder-dwelling S. haematobium, liver flukes or lung flukes.
Protocol
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison under Protocol no. V005717. The following protocol involves working in a Biosafety Level 2 (BSL2) facility for human pathogens, although none of the schistosome stages depicted in this protocol are infective to humans or other mammals. Follow institutional policies for handling Risk Group 2 (RG2) human pathogens.
NOTE: We use female Swiss-Webster mice (6-week old) due to their higher susceptibility to infection10, thereby yielding larger egg burdens. Tucker et al. describe the mouse and snail infection protocols used in this model system11. The Table of Materials lists all of the specific equipment, materials, reagents and animal sources needed to carry out this experimental protocol.
1. Processing of infected livers
- Euthanize mice, 6-7 weeks post-infection, by CO2 asphyxiation (rate of 10-30% of the chamber's volume per minute). Monitor mice for cessation of respiration and heartbeat.
NOTE: Typically, up to 20 mice may be processed at a given time. - After transferring euthanized mice to a biosafety cabinet, place mice on their backs and spray their ventral surfaces with 70% ethanol. Let soak for 1 min.
- Pinch and pull up skin of the lower abdomen and cut across the base of the pinched skin closest to the abdomen with sterile surgical scissors. Pull the cut skin anteriorly (i.e., toward the head) to reveal the liver. Note that the liver should be enlarged and speckled due to an egg-induced granulomatous response (Figure 2A).
- Remove the liver and place it in a beaker containing 200 mL of sterile 1.2% NaCl solution containing pen/strep.
- Repeat steps 1.3 and 1.4 with remaining mice.
NOTE: Twenty mice are usually processed in a single harvest. - Place the livers in a sterile Petri dish and remove non-liver fat/connective tissues with sterile forceps.
- Transfer trimmed/cleaned livers to a sterile 250-mL centrifuge bottle containing ~200 mL of sterile 1.2% NaCl solution with pen/strep.
- Vigorously shake the bottle containing the livers and, using a sterile disposable pipette, remove the excess surface foam/floating debris. Slowly pour off the remaining saline solution into a container or sink containing 1% bleach (disinfectant).
- Add ~200 mL of fresh saline solution to the livers and repeat step 1.8 three more times.
2. Liver extraction and miracidial isolation
- Place the livers into a small sterile stainless-steel blender cup and add ~2 mL of 1.2% NaCl solution (Figure 2B).
- Cover the cup with foil and a petri dish to prevent spills, and blend for 1 min by alternating low and high speed (20 s low speed/10 s high speed; repeat) (Figure 2C).
- Evenly distribute the blended liver suspension into sterile 250-mL centrifuge bottles (for 20 livers use 4 bottles).
- Add sterile saline with antibiotics to ~200 mL total volume/bottle. Weigh/balance bottles prior to centrifugation.
- Centrifuge the blended livers at 290 x g for 15 min at 4 oC. Gently pour off supernatants into a container with bleach prior to discarding.
- Repeat steps 2.4-2.5 once. Be sure to thoroughly resuspend liver sediment prior to centrifugation.
- After discarding the last saline wash (Step 2.6), add 200 mL sterile pond water with pen/strep to the pelleted liver tissue, shake to resuspend sediment, and transfer the suspension to a sterilized 1-L volumetric flask that has been completely covered with aluminum foil, except for the top 3 cm of the neck (Figure 2D).
NOTE: Use two bottles (= 10 livers) per 1-L flask. Pond water simulates the natural freshwater environment needed to stimulate miracidial hatching from eggs. - Using sterile pond water, fill up the flask to approximately 3 cm above the foil covering on neck. Then, without delay (as miracidia soon begin to hatch), remove residual foam and liver tissue floating to the surface by quickly pipetting up ~12 mL of pond water containing debris and discarding it into a separate discard tube. Replace with fresh sterile pond water.
- Repeat cleaning Step 2.8, three more times, checking for presence of miracidia in the discard tube. Cease repetition of cleaning step if miracidia appear in the wash solution.
- After the last wash, slowly add ~12 mL of warm sterile pond water (pre-warmed to 30 oC) to create a temperature gradient with clean, sterile warm water on top.
NOTE: This is best accomplished by slowly discharging water along the side of the flask neck to avoid mixing. - Place a small Petri dish cover over the flask opening and shine a bright light across the top of the flask above the foil cover to illuminate the upper surface of water.
NOTE: Typically, within 5-10 min, swimming miracidia, attracted to the light, will concentrate in large numbers within the first 2-3 cm of pond water (Figure 3A).
3. Miracidial harvesting and cultivation
- When miracidia have amassed at the surface after 10 min, using a sterile transfer pipet, remove ~8 mL of pond water and transfer to a sterile 15-mL centrifuge tube. Place the tube containing miracidia on ice.
- Add back 8 mL of warm sterile pond water along the inside of the volumetric flask and wait another 10 min.
- Using new tubes each time, repeat steps 3.1 and 3.2 three more times. After the 4th collection, keep the final tube on ice for 10 min to allow the miracidia to settle. This set of tubes comprises the first harvest.
- Centrifuge tubes containing miracidia at 290 x g for 1 min at 4 oC in a pre-cooled refrigerated centrifuge.
- Immediately remove most of the supernatant (~7 mL) being careful not to disturb the parasite pellet (Figure 3B).
- Pool all the miracidia from this first harvest into one tube, then rinse all of the original tubes with ~1 mL sterile pond water and add to the "pooled" tube.
- Allow parasites to settle on ice for ~5 min, and again centrifuge the parasites at 290 x g for 1 min at 4 oC.
- Carefully remove the supernatant and add 6-8 mL sterile Chernin's balanced salt solution (CBSS+; see Table of Materials) containing pen/strep.
- Gently suspend the miracidia and aliquot them into a 24-well culture plate (1 mL per well), followed by rinsing the tube in 6-8 mL CBSS+ and adding 1 mL of rinse to each well. Incubate parasites at 26 oC under normoxic conditions. Concentrations of isolated miracidia will vary, but will usually average ~7000/mL.
- If parasites are still concentrating at the light source, repeat Steps 3.1 to 3.9 to continue harvesting late-hatching miracidia but increase the time interval between collections to 15-20 min.
NOTE: This new set of tubes represents the second harvest. However, because miracidial numbers typically are low, larvae from all tubes usually are pooled into a single culture well containing 2 mL CBSS+. - At 24 h post-harvest and cultivation, gently resuspend sporocysts in their wells by pipetting.
- Wait a few min to allow parasites to settle to the bottom of the wells. Once they have settled, carefully remove the supernatant (~1.5 mL), which will contain most of the ciliated epidermal plates shed by miracidia during larval transformation. This "transformation" supernatant may either be discarded or incorporated into follow-up studies of larval secretory products.
- Add 1.5 mL of fresh CBSS+ to each well and gently pipette to resuspend sporocysts. Repeat step 3.12 if further washing or changing to a different medium is desired.
Representative Results
The vast majority of miracidia typically will have been collected in the first two "harvests". Incubation of isolated miracidia in CBSS+ triggers the miracidium-to-sporocyst transformation process (Figure 4A-C). Within the first hour following transfer to culture wells containing CBSS+, the majority of miracidia cease swimming (Figure 4A). At 6 h in culture, miracidia are in the process of actively shedding their ciliated epidermal plates (Figure 4B). Coincident with this shedding process, larvae form their outer syncytial tegumental covering as they transform to primary sporocysts. Most of the parasites will have completed larval transformation, becoming fully-formed sporocysts, by 24-48 h post-cultivation in CBSS+ (Figure 4C). After 4 days of culture in CBSS+ alone, sporocyst survival may be expected to decrease to ~80%. However, sporocysts cultured for 1 day in CBSS+ and then transferred to complete Bge (cBge) medium for 3 days typically result in better survival rates and larvae with a more robust appearance (Figure 4D). Sporocyst viability generally remains stable between 4 and 7 days in cBge with continued increase in larval growth during this time in culture compared to CBSS+ alone.
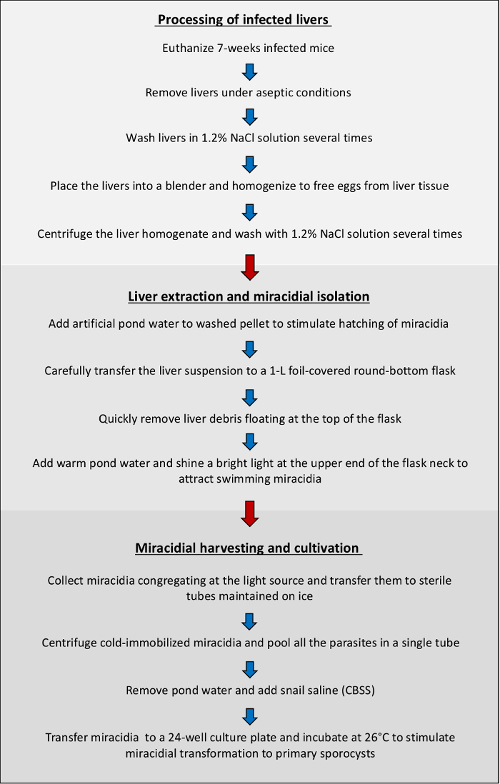
Figure 1. Overview of protocol workflow. Schematic summary of the major steps involved in the isolation and cultivation of miracidia derived from mice infected with the human blood fluke, S. mansoni. Please click here to view a larger version of this figure.

Figure 2: Processing of the infected livers. (A) Mouse liver heavily infected with S. mansoni eggs. Note the enlarged size, abnormal color and the presence of granulomas in the infected liver (right) compared to a normal uninfected liver (left). (B) Washed mouse livers after transfer to sterile stainless-steel blender cup. Livers were then blended for one minute using sterile foil and Petri dish to cover the cup, thus avoiding spills (C). After blending, the liver homogenate, containing eggs, was washed several times in saline and resuspended in sterile pond water, before being transferred to a 1-L volumetric flask covered with aluminum foil (D). Please click here to view a larger version of this figure.

Figure 3: Harvesting S. mansoni miracidia. Actively swimming miracidia concentrated by attraction to light at the top of the volumetric flask (A). At 10-min intervals, parasites are harvested by pipette and placed at 4 °C to immobilize larvae. Miracidia are then washed in CBSS+ by centrifugation and collected in a single tube (B) just before distribution to wells of a culture plate. CBSS+; Chernin's balanced salt solution (Table of Materials). Please click here to view a larger version of this figure.
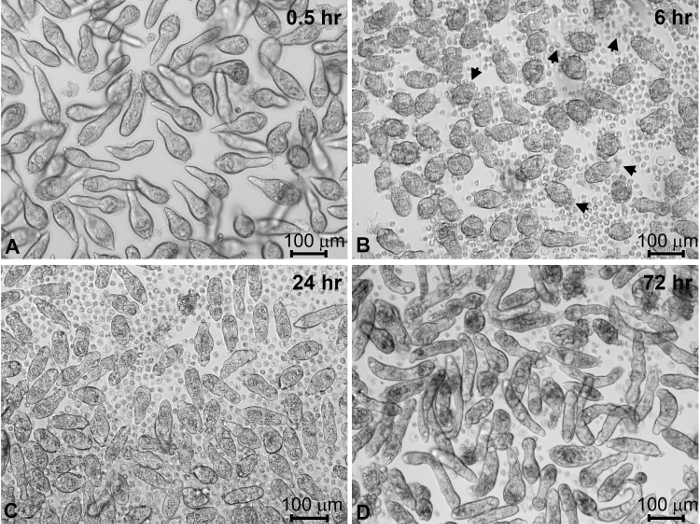
Figure 4: Miracidial transformation to primary sporocysts and their maintenance in vitro. S. mansoni miracidia and sporocysts placed into in vitro culture for different times post-harvesting. Newly harvested miracidia within 1 h of culture in CBSS+ (A). After 6 h of culture, miracidia have begun to shed their ciliated epidermal plates (arrows) as they transform to primary sporocysts (B). After 24 h in CBSS+ most of the miracidia have completely transformed to primary sporocysts (C). Sporocysts were transformed in CBSS+ for 24 h, then transferred and maintained in cBge medium for 3 days (D). Sporocysts generally appear more robust and have higher survival rates at 4 days post-cultivation in the presence of cBge, compared to CBSS+ alone. CBSS+, Chernin's balanced salt solution; cBge medium, complete B. glabrata embryonic cell medium (see Table of Materials). Please click here to view a larger version of this figure.
Discussion
Miracidia of S. mansoni, isolated and manipulated as described herein, can infect only the snail intermediate host, and therefore do not represent a human biohazard during this phase of larval development. However, to avoid accidental exposure/infection of snails, care should be taken to perform miracidial isolations in a different location from areas where susceptible Biomphalaria snail species may be present or maintained. A separate room, registered as BSL2 space, is highly recommended. In addition, all wash solutions used in the processing of livers and larval isolation should be treated with disinfectant (e.g., 1% bleach) before disposal.
It should be noted that surgical removal of livers from infected mice and their subsequent processing prior to blending/homogenization are performed in a biological safety cabinet (BSC) under aseptic conditions. However, for practical reasons (ease of manipulating bulk samples) all other procedures are done on a disinfected benchtop utilizing sterile containers (e.g., blender cups, flasks, bottles and tubes, transfer pipets, etc.) and sterile solutions containing antibiotics (saline, pond water, CBSS). Antibiotics are added to all solutions as a further precaution against introduced microbial contamination. In addition, during the miracidial isolation and harvesting steps (Protocol Sections 2 and 3), a small Petri dish cover is placed over the flask opening to minimize outside contamination during initial removal of foam/liver debris and subsequent transfer of concentrated miracidia from the flask to centrifuge tubes.
Typically, we harvest miracidia from 20 infected mouse livers in a single batch, from which we estimate larval yields of about 5000 larvae per liver. However, yields can vary considerably depending on the infection intensity of individual mice resulting in batch-to-batch variation in miracidial recoveries. Isolations can be carried out using fewer starting livers, although the method we have described is generally applicable for 10-20 livers. When 20 livers are processed, following the final resuspension of the liver pellets in sterile pond water (Protocol Section 2, Step 2.7), the suspensions should be distributed equally into two 1000-mL flasks (2 bottles/flask). If 10 livers or fewer are used, the original liver homogenates (Protocol Section 2, Step 2.3) can be distributed to 2 bottles and washed suspensions then transferred to a single volumetric flask for further processing. One or 2 livers also can be processed, but quantities (volumes) of extraction, wash and hatching solutions should be reduced proportionately, with final washed sediment in pond water being transferred to a 10-mL foil-covered volumetric flask to concentrate miracidia.
In order to maximize miracidial yields with a minimum of liver debris contamination, it is also important to perform the pond water washes (Protocol Section 2, Steps 2.7-2.9) as quickly and efficiently as possible. Once eggs are exposed to pond water, miracidial hatching occurs soon thereafter, although the time before larval appearance at the light source in the flask neck can vary from 5-20 min after hatching commences. To limit loss of parasites during pond water cleaning steps it is crucial to visually check each pond water wash for early arriving miracidia in the upper water layer of the flask's neck. In addition, once miracidia are transferred to 15-mL collection tubes and are placed on ice, they will cease swimming activity and settle to the tube bottom. However, after centrifugation to pellet larvae (Section 3, Steps 3.4-3.5), miracidia will again start to swim if water is allowed to warm. Therefore, it is important to remove the pond water quickly, but without disturbing the pellet or pipetting up swimming parasites.
As previously mentioned, yields of isolated miracidia can vary depending on the number of adult worm pairs present in individual mice (infection intensity) and consistency of initial infection protocols. Miracidia are strongly phototropic and the majority (80-90%) will concentrate at the light source over the first harvest period. The remaining 10-20% of miracidia will continue to hatch and more slowly accumulate during the second harvest period. Most (>95%) miracidia introduced into culture and maintained at 26 oC in CBSS+ medium will have transformed to primary sporocysts within 24-48 h of cultivation. At this time, larval viability typically is >90%. After 4 days in CBSS+ alone viability drops considerably (70-80%). However, switching the culture medium from CBSS+ to complete Bge medium 24 h following initial CBSS cultivation results in higher survival rates and more robust growth. We routinely maintain short-term sporocyst cultures under normoxic conditions. However, sporocysts are quite sensitive to oxygen levels, and especially to reactive oxygen species that can inflict deleterious oxidative damage7. C.J. Bayne4 and others have reported that the overall health and viability of sporocysts in vitro are significantly improved when cultures are maintained under hypoxic conditions. Lowered oxygen levels may be obtained by gassing (with nitrogen) individual closeable flasks, or by nitrogen-enrichment of the gas phase of the incubator4.
As cited above, methods for axenic isolating and culturing of schistosome miracidia and sporocysts have been described previously. It was recognized early on that miracidial phototropism could be used to attract and concentrate swimming larvae and that isolated miracidia could be rapidly immobilized in hyperosmotic saline solutions (between 120-160 mOs/Kg)8. It was further observed that maintenance in saline solutions resulted in the transformation of miracidia to primary sporocysts, the first intramolluscan larval stage9,12. Complex media also have been formulated to support longer-term in vitro growth and development of the S. mansoni sporocyst stages9,13,14,15, as well as cells derived from the snail host Biomphalaria glabrata16. These formulations incorporated various commercial media, and may have been supplemented with amino acids, salts, lipids, reducing agents, and sugars. Complete media formulations also included heat-inactivated fetal bovine or horse serum at various concentrations. We have found that proper heat-inactivation (HI) was crucial to sporocyst survival and long-term development in vitro. We found that incubation of serum (thawed, 100-mL bottles) in a 60 oC waterbath for 60 min, with gentle mixing every 10 min for uniform heating was most effective. In addition, we usually test several lots of serum from a prospective supplier for larval culture compatibility. Finally, the establishment of the first (and currently only) authentic molluscan cell line, the B. glabrata embryonic (Bge) cell line by Hansen16 was a major breakthrough that significantly facilitated advances in larval schistosome cultivation efforts. Isolated S. mansoni miracidia, when placed into co-culture with Bge cells developed from primary to secondary/daughter sporocysts17, and eventually to the final cercarial stage18,19. Thus, continuous in vitro cultivation of the entire snail phase of the S. mansoni life cycle has been achieved. The Bge cell medium used in our culture system supports both sporocyst growth/development and maintenance of the Bge cell line.
In summary, we have described and visually illustrated a detailed protocol for the mass axenic isolation and cultivation of the free-swimming miracidial stage of S. mansoni. These miracidia, when subjected to appropriate culture conditions, are capable of developing to successive primary and secondary sporocyst generations, and eventually cercariae. With the development and continual refinement of tools currently available for manipulating genes and gene expression in vitro through transgenic, RNA interference and genome editing (e.g., CRISPR/Cas9) approaches, it is envisioned that efficient methods for isolation and cultivation of miracidia from diverse trematode species20 will continue to be needed in order to procure materials for further advancement of research in this field. This method nicely complements a previously described protocol for isolating and culturing free-swimming cercariae of S. mansoni to facilitate in vitro transformation to the mammalian schistosomula stage21, for eventual cultivation to ovigerous adult worms22.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
Funded in part by NIH grant RO1AI015503. Schistosome-infected mice were provided by the NIAID Schistosomiasis Resource Center at the Biomedical Research Institute (Rockville, MD) through NIH-NIAID Contract HHSN272201000005I for distribution through BEI Resources.
Materials
| Chernin's balanced salt solution (CBSS+) | For 1L of solution | ||
| 2.8 g sodium chloride | Fisher Scientific | S271-3 | Dissolve salts, except calcium chloride, and |
| 0.15 g of potassium chloride | Sigma-Aldrich | P5405 | sugars in 800 mL ddH2O |
| 0.07 g sodium phosphate, dibasic anhydrous | Fisher Scientific | S374-500 | Dissolve calcium chloride separately in 200 |
| 0.45 g magnesium sulfate heptahydrate | Sigma-Aldrich | M1880 | mL ddH2O |
| 0.53 g calcium chloride dihydrate | Mallinckrodt | 4160 | Slowly add calcium soln to the salt/sugar soln with |
| 0.05 g sodium bicarbonate | Fisher Scientific | S233-3 | with constant mixing |
| 1 g glucose | MP Biomedicals | 152527 | Adjust to pH 7.2 and filter sterilize using a |
| 1 g trehalose | Sigma-Aldrich | T0167 | 0.22 µm disposable bottle-top filter |
| 10 mL 100X penicillin/streptomycin | Hyclone | SV30010 | Add filtered penicillin and streptomycin soln prior use |
| Incomplete Bge medium (Ibge) | For 900 mL solution | ||
| 220 mL Schneider’s Drosophila medium modified | Lonza | 04-351Q | Mix Schneider's medium with 680 mL ddH2O |
| 4.5 g lactalbumin enzymatic hydrolysate | Sigma-Aldrich | L9010 | Add lactalbumin hydrolysate and galactose |
| 1.3 g galactose | Sigma-Aldrich | G0625 | Adjust to pH 7.2 and filter sterilize using a 0.22 µm pre-sterilized disposable bottle-top filter |
| Complete Bge medium (cBge) | For 100 mL of solution | ||
| 90 mL Incomplete Bge medium | To heat-inactivate FBS: Incubate thawed FBS in | ||
| 9 mL heat-inact. fetal bovine serum (FBS) (Optima) | Atlanta Biologicals | S12450 | waterbath at 60°C for 1 hr while gently |
| 1 mL 100X penicillin/streptomycin | Hyclone | SV30010 | swirling the bottle every 10 min Aliquot heat-inactivated FBS into 15-mL tubes and store at -20°C. Mix medium + FBS and filter sterilize using a 0.22 µm pre-sterilized disposable bottle-top filter Add penicillin and streptomycin prior to use |
| Pond water (stock solution) | 1L of stock solution | ||
| 12.5 g calcium carbonate | Fisher Scientific | C64-500 | Mix all salts in 1L of ddH2O |
| 1.25 g magnesium carbonate | Fisher Scientific | M27-500 | Note that the salts will not have completely |
| 1.25 g sodium chloride | Fisher Scientific | S271-3 | dissolved. Shake vigorously to suspend |
| 0.25 g potassium chloride | Sigma-Aldrich | P5405 | salts prior to making the working soln |
| Pond water (working solution) | 1.5L of solution | ||
| 0.8 mL stock solution pond water (shake prior use) in | Mix stock to ddH2O | ||
| 1500 mL of ddH2O | Sterilize pond water by autoclaving (slow cycle) | ||
| 1.5 mL of 100X penicillin/streptomycin | Hyclone | SV30010 | Add penicillin and streptomycin prior use |
| Saline solution (1.2% NaCl) | 1.5L of solution | ||
| 18 g sodium chloride in 1500 mL of ddH2O | Fisher Scientific | S271-3 | Autoclave saline solution to sterilize |
| 1.5 mL of 100X penicillin/ streptomycin | Hyclone | SV30010 | Add penicillin and streptomycin prior use |
| Additional equipment and material: | |||
| 7-L mouse euthanizing chamber | Following approved IACUC protocol no. V001551 | ||
| Mice | Taconic Biosciences | Swiss-Webster, female, 6-wk old, murine | |
| CO2 tank and regulator | pathogen-free | ||
| 24-well tissue culture plate | TPP | 92424 | |
| 1-L volumetric flasks | |||
| Light source (150W) | Chiu Tech Corp | Model F0-150 | |
| Centrifuge, refrigerated, swinging bucket | Eppendorf | Model 5810R | |
| Centrifuge bottles (250 mL) | Nalgene | ||
| 15-mL centrifuge tubes, sterile | Corning | 430053 | |
| Sterile disposable transfer pipets | Fisher Scientific | 1371120 | |
| 0.22 µm pre-sterilized disposable bottle-top filter | EMD Millipore | SCGPS05RE | |
| Stainless steel blender | Waring Commercial | Model 51BL31 | |
| Blender cup, 100 mL capacity | Waring Commercial | ||
| Inverted compound microscope | Nikon Instruments | Eclipse TE300 |
Referenzen
- Colley, D. G., Bustinduy, A. L., Secor, W. E., King, C. H. Human schistosomiasis. Lancet. 383, 2253-2264 (2014).
- WHO. Schistosomiasis: number of people treated worldwide in 2013. Wkly. Epidemiol. Rec. 5 (90), 25-32 (2015).
- Yoshino, T. P., Gourbal, B., Théron, A., Jamieson, B. G. M. Schistosoma sporocysts. Schistosoma: biology, pathology and control. , 118-148 (2017).
- Bayne, C. J. Successful parasitism of vector snail Biomphalaria glabrata by the human blood fluke (trematode) Schistosoma mansoni: a 2009 assessment. Mol. Biochem. Parasitol. 165 (1), 8-18 (2009).
- Yoshino, T. P., Coustau, C., Toledo, R., Fried, B. Immunobiology of Biomphalaria-trematode interactions. Biomphalaria snails and larval trematodes. , 159-189 (2011).
- Coustau, C., et al. Advances in gastropod immunity from the study of the interaction between the snail Biomphalaria glabrata and its parasites: A review of research progress over the last decade. Fish Shellfish Immunol. 46 (1), 5-16 (2015).
- Bayne, C. J., Hahn, U. K., Bender, R. C. Mechanisms of molluscan host resistance and of parasite strategies for survival. Parasitology. 123, S159-S167 (2001).
- McMullen, D. B., Beaver, P. C. Studies on schistosome dermatitis. IX. The life cycles of three dermatitis-producing schistosomes from birds and a discussion of the subfamily Bilharziellinae (Trematoda: Schistosomatidae). Amer. J. Hyg. 42, 128-154 (1945).
- Voge, M., Seidel, J. S. Transformation in vitro of miracidia of Schistosoma mansoni and S. japonicum into young sprocysts. J. Parasitol. 58 (4), 699-704 (1972).
- Eloi-Santos, S., Olsen, N. J., Correa-Oliveira, R., Colley, D. G. Schistosoma mansoni: mortality, pathophysiology, and susceptibility differences in male and female mice. Exp. Parasitol. 75 (2), 168-175 (1992).
- Tucker, M. S., Karunaratne, L. B., Lewis, F. A., Freitas, T. C., Liang, Y. S. Schistosomiasis. Curr Proto Immunol. 103, 19.1:19.1.1-19.1.58 (2013).
- Basch, P. F., DiConza, J. J. The miracidium-sporocyst transition in Schistosoma mansoni: surface changes in vitro with ultrastructural correlation. J. Parasitol. 60 (6), 935-941 (1974).
- Hansen, E. L. Secondary daughter sporocysts of Schistosoma mansoni: their occurrence and cultivation. Ann. N. Y. Acad. Sci. 266, 426-436 (1975).
- Stibbs, H. H., Owczarzak, O., Bayne, C. J., DeWan, P. Schistosome sporocyst-killing amoebae Isolated from Biomphalaria glabrata. J. Invertebr. Pathol. 33, 159-170 (1979).
- DiConza, J. J., Basch, P. F. Axenic cultivation of Schistosoma mansoni daughter sporocysts. J. Parasitol. 60 (5), 757-763 (1974).
- Hansen, E. L., Maramorosch, K. A cell line from embryos of Biomphalaria glabrata (Pulmonata): Establishment and characteristics. Invertebrate tissue culture: Research applications. , 75-97 (1976).
- Yoshino, T. P., Laursen, J. R. Production of Schistosoma mansoni daughter sporocysts from mother sporocysts maintained in synxenic culture with Biomphalaria glabrata embryonic (Bge) cells. J. Parasitol. 81 (5), 714-722 (1995).
- Ivanchenko, M. G., et al. Continuous in vitro propagation and differentiation of cultures of the intramolluscan stages of the human parasite Schistosoma mansoni. Proc. Natl. Acad. Sci. USA. 96, 4965-4970 (1999).
- Yoshino, T. P., Bayne, C. J., Bickham, U. Molluscan cells in culture: primary cell cultures and cell lines. Can. J. Zool. 91, 391-404 (2013).
- Coustau, C., Yoshino, T. P. Flukes without snails: Advances in the in vitro cultivation of intramolluscan stages of trematodes. Exp. Parasitol. 94, 62-66 (2000).
- Milligan, J. N., Jolly, E. R. Cercarial transformation and in vitro cultivation of Schistosoma mansoni schistosomules. J. Vis. Exp. (54), e3191 (2011).
- Basch, P. F. Cultivation of Schistosoma mansoniin vitro. II production of infertile eggs by worm pairs cultured from cercariae. J. Parasitol. 67 (2), 186-190 (1981).
