This protocol was approved and performed in accordance with IACUC of University of Toronto.
1. Design and cloning of pooled CRISPR libraries
- Select 4-5 sgRNAs targeting mouse genes of interest from resource such as the Broad Institute sgRNA designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) or CHOPCHOP server (https://chopchop.cbu.uib.no). Select an equal number of non-targeting sgRNAs from e.g., Sanjana et al.9 to generate an equally sized non-targeting controls gRNA library.
- While constructing the sgRNA libraries, make sure there is enough coverage for each sgRNA in the targeted organ system. For the mouse skin, the epidermis at E9.5 is a single layer that contains ~150,000 cells, most of which have stem cell capacity4. If lenti-viral transduction results in 15-20% infectivity, only 18,000-24,000 cells will be transduced at E9.5. Scale the experiment accordingly.
- For cloning and amplification of these sgRNA libraries, add BsmBI enzyme restriction sites as well as primer binding DNA sequences to 5’ and 3’ end of the sgRNA and order the resulting full length oligos as pooled oligonucleotide chip (Figure 1A).
- To multiplex different libraries on one chip, add library specific primer sequences.
- Using the appropriate primer pairs, amplify each library separately from the pooled oligo chip. In a PCR tube, mix 25 µL of 2x polymerase master mix, 20 µL of DNase/RNase free water, 5 ng of oligo chip DNA, 2.5 µL of appropriate forward primer and 2.5 µL of appropriate reverse primer. Use 12-15 cycles and amplify with 98 °C denaturation, 63-67 °C annealing, 72 °C extension parameters for each cycle in the PCR machine.
- Run PCR product on a 2.5% agarose gel and purify ~100 bp PCR product using a gel DNA clean-up kit.
- Prepare the backbone plasmid.
- Digest 5 µg of the Cre-recombinase containing pLKO-Cre stuffer v3 plasmid with 2 µL of BsmBI in 50 µL reaction mix for 1 h at 55 °C, followed by 1 µL of alkaline phosphatase incubation for 45 min at 37 °C according to manufacturer’s instructions.
NOTE: Cre-recombinase containing pLKO-Cre stuffer v3 plasmid is a lenti-viral based plasmid that contains Cre-recombinase enzyme to remove Lox-Stop-Lox cassette in mouse cells and also has a U6 promoter to drive the expression of sgRNA and tracrRNA. A version with Cas9 and without Cas9 can be used and available on Addgene #158030 and #158031. - Run the digested DNA on a 1% agarose gel and purify the 7 kb linearized vector band using a gel DNA clean-up kit. Of note, a 2 kb stuffer band should also be visible indicating a successful digest.
- Digest 5 µg of the Cre-recombinase containing pLKO-Cre stuffer v3 plasmid with 2 µL of BsmBI in 50 µL reaction mix for 1 h at 55 °C, followed by 1 µL of alkaline phosphatase incubation for 45 min at 37 °C according to manufacturer’s instructions.
- Set up the ligation reaction to generate a sgRNA plasmid library.
- Mix 1 µg of purified vector and 30 ng of purified PCR insert with 2 µL of BsmBI, 5 µL of T4 DNA ligase, 10 µM ATP and 1x buffer specific to BsmBI. Incubate the ligation mix overnight at 37 °C. Use an additional tube containing all the above materials except the PCR insert as a negative control.
- Next day morning, purify the ligation mix using the oligo clean-up kit and elute in 7 µL of RNase/DNase free water.
- Electroporate the library into competent cells.
- Add 2 µL of eluted sgRNA library or negative control ligation mix to 25 µL of thawed electrocompetent cells in pre-chilled cuvettes (1.0 mm) on ice. Electroporate following the manufacturer’s protocol (10 µF, 600 Ohms, 1800 Volts). To the cuvette add 975 μL of recovery medium (or SOC medium) within 10 s of the pulse.
- Transfer electroporated cells to a culture tube and incubate for an hour at 37 °C in a bacterial shaking incubator at 300 rpm.
- Estimate transformation efficiency and the library coverage per sgRNA.
- Prepare a 100-fold dilution by transferring 10 μL of the transformation reaction containing cells electroporated with sgRNA library or negative control ligation to 990 μL of Recovery Medium and mix well.
- Plate 10 μL of the diluted transformation mix onto a pre-warmed 10 cm LB + ampicillin (100 µg/L) agar plate. This results in a 10,000-fold dilution of the transformants and use this plate to calculate the transformation efficiency. Perform incubation of the plates at 30 °C for 14–16 h.
- Plate the rest of the transformation reaction by spreading 100 μL of recovered cells on each plate of a total of 10 pre-warmed 15 cm LB + ampicillin agar plates. Incubate the plates for 14–16 h at 30 °C. Of note, growth at 30 °C minimizes recombination between the two long-terminal repeats within the viral plasmid.
- To assess the cloning success, calculate the transformation efficiency. Count the number of colonies on the dilution plate containing transformants from sgRNA library or negative control ligation (10 cm plates, step 1.8.2). Negative control mix should not have any or only very few colonies. To obtain the total number of colonies, multiply the number of colonies by 10,000.
NOTE: The counted total number of colonies represents a library coverage which should be minimum of 200x colonies per sgRNA.- Ensure that a library with 2000 sgRNAs will has at least 400,000 colonies. In case there are not enough colonies, repeat and set up more electroporation.
- Quality control: From the dilution plate, pick 20 colonies and add each colony to an individual culture tube containing 3 mL of LB media + ampicillin. Incubate all 20 tubes overnight at 30 °C shaking at 250 rpm. Purify the plasmid DNA using mini-prep kit according to manufacturer’s instructions and Sanger sequence all the 20 plasmid DNA samples to verify each sample has a different sgRNA sequence using the U6 primer (5’-GAG GGC CTA TTT CCC ATG ATT CC-3’).
- Harvest the colonies from each 15 cm plate.
- Add 7 mL of LB medium, then scrape the colonies off the LB Agar plate with a cell spreader. Use a 10 mL pipette to transfer the cells into a 2 L sterile conical flask. Repeat for all plates and pool bacteria in the 2 L flask. Incubate the flask for 2-3 h shaking at 30 °C. Centrifuge the culture and collect the pellet.
- Purify plasmid DNA using a maxi- plasmid purification kit.
- Additional quality control: Use 1 ng of maxi-prep sgRNA library plasmid DNA and run a PCR reaction using Next-Generation sequencing primers according to manufacturer’s instructions. The representation of all sgRNAs in the library can be verified by deep sequencing platform.
2. Production of high titer lentivirus suitable for in vivo transduction
NOTE: Perform all steps in this section of the protocol in a BSL2+ facility in a Class II, Type A2 biosafety cabinet. 293FT and especially the 293NT packaging cells allow for higher virus production. Use low passage (<p20) HEK293T, 293FT or 293NT cells for transfections. Prewarm all media to 37 °C. Never allow HEK293T, 293FT or 293NT cells to become confluent while subculturing. Grow 293FT in the presence of G418 to maintain the expression of the SV40 large T-antigen.
- Prepare viral packaging cells.
- On Day 1, plate HEK293T cells on 6 poly-L-lysine coated 15 cm plates at 35% confluency in growth media (DMEM + 10% FBS + 1% Penicillin-Streptomycin antibiotic solution (w/v)). Incubate cells overnight at 37 °C, 5% CO2.
- Once the plated cells are 70-80% confluent and evenly spread, replace growth media with 25 mL of serum and antibiotic-free DMEM media.
- Add media slowly to sides of flasks when feeding, as HEK293T and 293NT cells can detach easily from tissue culture plastic
- Prepare transfection mix.
- Add 65 µg of lentiviral packaging plasmids psPAX2, 43 µg of VSV-G expressing envelope plasmid pMD2.G, 65 µg of sgRNA library plasmid and 240 µL of 1 mg/mL polyethyleneimine (PEI) to 6 mL of DMEM and vortex.
NOTE: Always use an unopened or very recently opened bottle of DMEM for the transfection, as the pH at this step is critical, and DMEM becomes more basic over time once opened. - Incubate for 15 min at room temperature. This transfection mix is enough for cells plated on 6 x 15 cm plates with 870 cm2 surface area. Scale the transfection mix according to the requirements of the experiment.
- Add 65 µg of lentiviral packaging plasmids psPAX2, 43 µg of VSV-G expressing envelope plasmid pMD2.G, 65 µg of sgRNA library plasmid and 240 µL of 1 mg/mL polyethyleneimine (PEI) to 6 mL of DMEM and vortex.
- Transfection of viral packaging cells.
- After 15 min of incubation, transfect each of the 15 cm plates by adding 1 mL of the transfection mixture dropwise throughout the plate. Incubate at 37 °C, 5% CO2 for 8 h.
- After incubation, remove the serum-free media containing the transfection mix and add 30 mL of regular culture media to the 15 cm plates and culture for 48 h at 37 °C, 5% CO2.
- After 48 h, collect the viral supernatant and filter through 0.45 μm PVDF filter. Keep 1 mL of the viral supernatant and store at -80 °C for quality control and viral titering.
- Concentrate the viral supernatant by sucrose-gradient centrifugation in high-speed centrifuge using SW28 rotor head.
- Prime ultracentrifuge tubes by washing them with 70% ethanol, followed by 3 rinses with PBS. Evenly distribute approximately 30 mL of the viral supernatant into each of the 6-ultracentrifuge tube.
- Gently pipette to the bottom of each tube 4 mL of the 20% sucrose solution (20 g of sucrose in 100 mL of PBS). Place the six ultracentrifuge tubes into the six SW28 swing buckets and precisely balance each opposing pair of buckets by adding or removing viral supernatant.
- Centrifuge viral supernatant at 4 °C for at least 2 h and up to 4 h at 80,000 x g and 4 °C. Once the centrifugation is done, carefully discard the supernatant.
- Drain the remaining supernatant by placing ultracentrifuge tubes upside down onto sterile soft paper towel for at least 2 min and wipe off any droplets of supernatant inside the tube using a soft paper towel to get rid of any residual medium. A small grayish white pellet might be visible at the bottom of the tube.
- Add 20-25 μL cold, fresh PBS to each tube. Seal tubes with paraffin film and incubate tubes upright on ice with gentle shaking on an orbital platform shaker for ~2 h.
- Using a 20 μL pipette, carefully dislodge and resuspend the pellet of the first tube, being careful not to form bubbles. Transfer the medium to the next tube and repeat the process till all the virus pellets have been dislodged and combined into one tube of approximately 120-150 μL volume. Incubate this high-titer viral suspension for another ~2 h on ice with gentle shaking.
- Transfer the viral suspension into a 1.5 mL microcentrifuge tube and spin the tube at 4 °C in a refrigerated and pre-cooled table-top centrifuge at 12,000 x g for 2 min. Aliquot the clear viral supernatant carefully as 10 μL aliquots in separate 0.2 mL tubes and store at -80 °C. Discard the pellet at the bottom of the tube as this will clog the needle during embryonic injections.
- Titration of pooled CRISPR lentivirus library
NOTE: The resulting viral suspension should yield an approximate 2,000-fold concentration and the resulting virus solution and should have viral titer of 107-109, which is good enough for more than 100 E9.5 surgeries described below.- Seed R26-Lox-STOP-Lox-tdTomato mouse embryonic fibroblasts (MEFs) or R26-Lox-STOP-Lox-tdTomato primary keratinocytes or any other Cre-reporter cell line into a 2 x 6 well plate.
- Once cell reach ~40% confluency, cells are ready for transduction. At that time, determine the number of cells within one well to enable calculation of viral titer later. Change the media of the Cre-reporter cells to growth media supplemented with 10 µg/mL polybrene (=hexadimethrine bromide).
- Dilute 1 μL of concentrated virus with 2 mL of growth media. Add 0, 10, 50, 200 µL of diluted viral suspension and 50, 200 µL of un-concentrated virus from step 2.4 and mix plates and incubate over night at 37 °C, 5% CO2.
- Remove growth media containing virus on the next day, wash cells with PBS and replace with normal growth media. The supernatant at this point is still considered viral waste and must be disposed in accordance to institutional BSL2 regulations.
- Approximately 36-48 h after transduction, assess Cre-mediated recombination of the Lox-STOP-Lox cassette and expression of tdTomato by FACS. Calculate viral titer. Calculate colony forming units (cfu)/mL using the following formula:
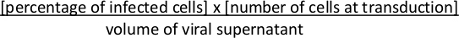
NOTE: Comparison between the concentrated viral suspension with its unconcentrated viral supernatant will allow to estimate the success of (1) viral production as well as (2) viral concentration. Alternatively, the viral titer can be estimated using qRT-PCR method10. Large-scale production and concentration of lentivirus can also be performed using a two-step concentration with a cartridge concentration followed by ultracentrifugation as previously described2.
3. Ultra-sound guided surgery and injection
NOTE: This technology was adapted from4,11. Microinjection geared towards transduction of the surface epithelium must be performed at embryonic day E9.5, when the surface ectoderm consists of a single layer and before formation of the periderm starting at E10, which would prevent transduction of this basal layer. Preferably set up mice on Friday, so that the first possible day with E9.5 embryos is the following Monday. Use Rosa26-Lox-STOP-LOX-Cas9-GFP mice (Jackson Laboratory #024857) for optimal CRISPR/Cas9 efficiency12.
- Set up appropriate male and female mice for timed pregnancies 10 days before the desired surgery date.
- Plug-check mice over the following days to identify potentially pregnant mice. Confirm the pregnancy the day before the planned surgery (i.e., E8.5) using ultrasound.
- Prepare the modified Petri dish.
- Using the double-sided sticky tape, glue the silicone membrane onto the bottom of the Petri dish covering the circular opening. Using sharp scissors, cut a 2 x 10 mm oval opening in the silicone membrane.
- Preparation of the microinjection system
- Prepare injection needles from a thick-walled glass capillary using a micropipette puller with the following settings: Pressure = 200; Heat = 769; Pull = 0; Velocity = 140; Time = 100.
- Using fine scissors to cut off the needle tip at the level where its diameter is ~30 μm. Bevel the needle using needle sharpener to 20° on a fine-grade abrasive plate with regular wetting for 20 min.
- Sterilize the microinjection needle by pushing 70% ethanol using a 10 mL syringe with a 26 G1/2 needle. Using a 10 mL syringe and 26 G1/2 needle loaded with mineral oil, backfill the microinjection needle. Ensure that there are no bubbles in the microinjection needle.
- Mount the microinjector needle on the micromanipulator according to the manufacturer instructions.
- Load the needle with viral sgRNA library.
- Pipette 10 μL of viral suspension onto a parafilm fixed onto a flat surface.
- Using micromanipulator, expel the mineral oil. The presence of a small volume of oil in the needle will prevent the lentivirus from contacting the piston. Load about 4-5 μL viral suspension by slow aspiration without any air bubbles.
- Anesthetize pregnant mouse using isoflurane induction chamber. Set the oxygen regulator to 1 L/min and isoflurane vaporizer to 2%. To determine if the animal is anesthetized, check by toe pinch after ~3-5 min in isoflurane induction chamber.
- Place the anesthetized animal ventral side up on the heated surgery stage (40 °C) and apply surgery tapes to hold the paws in place with the nose-cone anesthesia tube for constant supply of isoflurane and oxygen until the surgery is completed.
- Confirm E9.5 pregnancy if it was not done at E8.5.
- Apply a small quantity (~ ½ tsp) of a hair removal cream to the abdomen and spread it over a 3 x 3 cm area using a cotton-tipped applicator.
- After 2-3 min, gently remove the cream with gauze or tissue and wipe the area clean using PBS and 70% ethanol.
- Using the ultrasound probe and gel, check the pregnant mouse for embryonic day E9.5. Of note, this can also be done the day before at E8.5 to save time on the actual surgery day.
- Remove ultrasound gel and wipe area clean using BPS and then 70% ethanol to disinfect.
- Inject 0.02 cc of Buprenorphine analgesic subcutaneously in the mouse dam.
- Surgically expose the uterus
- Using sterile forceps and scissors, make a vertical ~2 cm incision into the skin of the mouse.
- Using blunt forceps separate the skin around the incision from the peritoneum.
- Using sterile forceps and scissors, cut through the peritoneum.
- Using blunt forceps, gently pull out the left and right uterus horn and count the embryos.
- Re-insert most of the uterine horns but leave the distal end of one uterine horn containing 3 embryos exposed.
- Using blunt forceps, gently push and pull the uterus with the 3 embryos through the opening in the silicone membrane of the modified Petri dish.
- Stabilize the Petri dish using cubes of modeling clay.
- Stabilize the uterus with the three embryos inside the Petri dish using a silicone mold. Flatten the silicone plug at the side of the embryos using a sharp knife.
- Fill up the Petri dish with sterile PBS. Flush the silicon membrane on the bottom of the Petri dish with the pregnant dam’s belly thereby preventing any leakage.
- Move the ultrasound head into the Petri dish ~0.5 cm atop the top embryo and adjust the stage so that the amniotic cavity becomes clearly visible in the ultrasound view.
- Align the injector needle carefully into the modified Petri dish. Using the micromanipulator, place the tip of the needle within ~5 mm of the top embryo. Then toggle the needle back and forth and move into the plane where its tip appears the brightest in the ultrasound image.
- Using the micromanipulator push the needle through the uterine wall into the amniotic cavity.
- Inject lentivirus containing sgRNA library.
- Pre-program the injector to 62 nL and slow injection speed. The microinjector can be programmed to inject specific volumes at a slow or fast speed.
- Press the Inject button 8 times for a total of 496 nL per embryo. Adjust the volume to required needs and viral titer. A viral titer of 1 x 108 pfu/mL and 496 nL injection volume will result in approximately 30% infectivity.
- Repeat the same procedure for other two embryos.
- Lift the ultrasound head and remove the silicone plug.
- Gently push the 3 embryos into the mouse abdomen using a sterile cotton swap.
- Gently pull out the next 3 embryos and repeat the injection procedure until desired number of embryos are injected.
NOTE: Do not exceed 30 min of anesthesia as pregnant dams are much more likely to abort their embryos beyond that time. An experienced surgeon can inject up to 12 embryos within 30 min surgery.
- Lift the ultrasound head, remove the micromanipulator with the needle, aspirate the PBS and remove the petri dish. Using sterile wipes, absorb any PBS that might have accumulated in the abdominal cavity.
- Close the peritoneal incision using absorbable sutures. Use two staples to close the incision in the abdominal skin.
NOTE: While performing ultra-sound guided in utero surgeries, utmost care should be taken to maintain a clean environment, maintain sterility as much as possible and avoid any possible contaminants. If more than one surgery must be done on the same day, it is important to sterilize the dissection instruments using a bead sterilizer and clean other apparatus that encounters mouse tissue cleaned with ethanol. This greatly increases the higher survival of the embryos post-surgery. The survival rate for the surgery method described here is consistently between 80-100%. - Post-surgery care.
- Allow the pregnant female to recover in a heated cage and observe for 15-30 min.
- Check vaginal canal for blood, which is an early sign of abortion and complications. For postoperative pain relief, deliver administer the analgesic twice a day for two days following surgery.
- Identify transduced embryos/newborn Rosa26-Lox-STOP-Lox-Cas9-GFP mice using a fluorescent dissecting microscope, a fluorescent protein flashlight and filter glasses or by genotyping for integrated viral particles in ear or tail clips.
NOTE: Mice are best identified using fluorescent microscope up to postnatal day 3 before hair starts growing. - To ensure enough coverage for the CRISPR library, calculate coverage based on the following parameters: E9.5 mouse surface ectoderm consists of ~150,000 cells; transduction of ~20% results in a minimal double infection rate (~1/10 double infected cells)4,5,11; at 20% infectivity, every embryo has 30,000 infected cells. Ensure that at least 100 individual cells are transduced with a given sgRNA. For example, a pool of 2000 sgRNAs requires 2 x 105 cells or 6-7 animals.
4. Deep sequencing procedure
- Upon experimental endpoint such as tumor formation or any other phenotype, sacrifice the mouse and harvest the tissue of interest. Use blood and tissue DNA extraction kit to purify genomic DNA following the manufacturer’s protocol. Before starting DNA extraction from tumors, clean the work bench with DNA Erase and work away from the lab area where plasmids are handled.
NOTE: It is important to generate a reference sample to allow for calculating sgRNA fold changes over time. Transduced embryos 3 days post infection or transduced cells (see step 2. 6) can serve as reference sample to determine sgRNA representation in the original library. - Use 100 ng – 1.5 μg genomic DNA as template in a 50 μL pre-amplification PCR 1 reaction using the outer primers listed in Table 1 and the PCR program outlined in Table 2. Set up enough reaction to yield desired coverage.
NOTE: Set up the barcoding PCR reaction in a separate dedicated area of the lab or in a tissue culture hood that is cleaned thoroughly by DNA erase to avoid any contamination. Run negative control for each PCR plate with water as template to make sure there is no contamination. - Pool all PCR 1 reaction and run 20 μL on a 1.5% control agarose gel to verify successful amplification of a 600 bp band.
- Use 5 μL of the pooled PCR1 reaction as template in a 50 μL of PCR 2 reaction using unique barcoded primer combination listed in Table 3 and the PCR program outlined in Table 4. Run 50 μL of PCR 2 reaction on a 2% agarose gel and purify the 200bp band using a gel extraction kit as per manufacturer instructions.
NOTE: The pre-amplification step is only required for amplification of a complex transduced tissue. If amplifying a single tumor that presumably developed form a clonal cell and thus contains just a single sgRNA, one can skip the pre-amplification PCR1 and proceed straight away with PCR2. - Quantify DNA concentration using fluorometric quantification and sent for deep sequencing to the sequencing facility (e.g., 1 million reads per tumor).
- Use Bowtie v1.2.3, which only allows ungapped alignments, align sequenced reads to sgRNA library13. Create a bowtie index using bowtie-build command inputting the sgRNA sequences in fasta format. Map the reads using bowtie command with options -m 2 -v 1, which allows maximum two mismatches during read alignment and discards reads that align more than one library sgRNA sequence. Typically, more than 80% of the reads align under these parameters.
- Use MAGeCK count command to obtain read count table using the aligned file as input14. sgRNA count table can be used for downstream analysis such as, determining the differential sgRNA’s (MAGeCK) and finding essential genes (BAGEL).
Figure 1A shows the design of the oligonucleotides for multiplexing several custom CRISPR libraries in a cost-effective manner in a single 12k or 92k oligo chip. Once the sgRNAs (blue color coded) are selected, the oligonucleotides are designed with restriction sites (orange colored BsmBI) and library specific PCR primer pairs (green color coded). Several libraries can be designed by using unique combination of primer pairs for multiplexing in a single oligo chip. When PCR amplifying the libraries using a specific PCR primer pair, always include a water only negative control and run all the reactions on an agarose gel. The negative control lane should not have any bands at 100 bp, while the library amplified lane should have a single 100 bp band. If there is no band, make sure the PCR primer pair is selected appropriately. Figure 1B shows the quality control step of the cloned library and the viral production and concentration procedure. Care should be taken to preserve the equal representation of sgRNAs from cloning till transduction. Next-generation sequencing reads of PCR amplified DNA from plasmid and lenti-viral library transduced cells should show a high correlation. Any deviation must be analyzed carefully to examine whether the representation is lost before or after the lenti-virus preparation. If the sgRNA reads from plasmid DNA do not show equal representation of sgRNAs, then the viral preparation and concentration procedure must be repeated with care. If the loss of equal sgRNA representation occurred in plasmid DNA, then the entire cloning procedure must be repeated including PCR amplification of libraries from oligo chip with reduced amplification cycles. The success of the CRISPR library screening depends critically on equal representation of the sgRNAs present in the library.
Figure 2 shows the ultrasound-guided micro-injection set up for manipulating the E9.5 embryos in pregnant mouse. The entire set-up is placed under a biosafety level class II cabinet to maintain the sterile condition of the entire procedure and to avoid any infection of the pregnant mouse due to the surgical procedures. Care should be taken to avoid pressuring the uterine horns while injecting. The needle must be very sharp, so that the wound on the uterine wall and the amniotic membrane is minimal.
Figure 3 shows the results of the ultra-sound guided injection of lenti-virus carrying Cre-recombinase in E9.5 Lox-stop-Lox (LSL) Confetti mouse pups at postnatal day (P)0. Viral titer can be adjusted to get an appropriate transduction coverage of the epidermis. While high titer of virus would result in larger coverage of the mouse skin, it would also result in transduction of multiple sgRNAs into the same cell potentially confounding the outcomes. To reduce multiple transductions of a single cell, the infection rate should be kept <20%.
Figure 4 shows next-generation sequencing reads of PCR amplified DNA from plasmid and lenti-viral library transduced cells and also reads from 4 representative tumors. sgRNA guides targeting Adam10 and Ripk4 are enriched in the tumor samples (triangles and diamonds) compared to sgRNA presentation in plasmid pool or infected cells. Adam10 and Ripk4 function as tumor suppressors5. Several hundred tumors can be multiplexed by assigning unique barcodes to each sample and deep sequenced as outlined in the protocol.
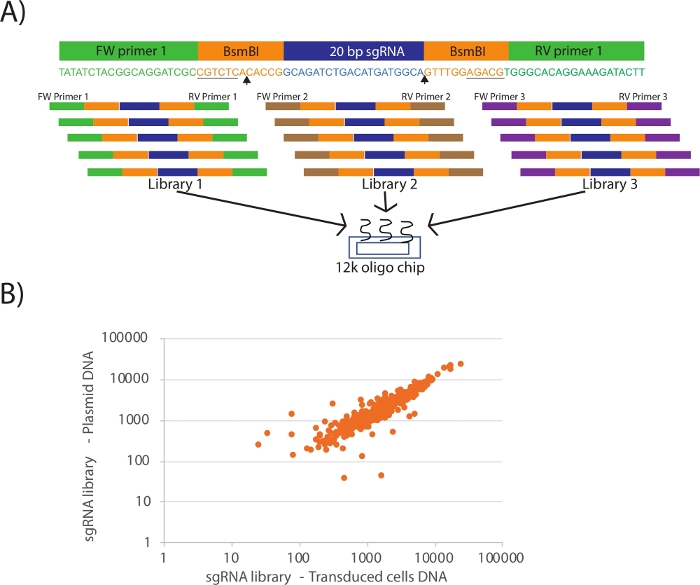
Figure 1: Cloning of targeted CRISPR library. (A) Schematic representing the design of oligonucleotides for the 12 k or 92 k custom oligo chip. BsmBI (or other compatible) restriction sites (orange color coded and underlined) were added on each side of the sgRNA. Arrow heads indicate the BsmBI cut site. For each library, a unique pair of PCR primers (green, brown and purple color coded) were added, so that it can be specifically amplified using PCR from the pooled chip. Several custom libraries can be multiplexed up to 12k or 92k oligos. (B) Graph showing sgRNA representation from a CRISPR library in plasmid DNA versus DNA from cells transduced with the same lentiviral library. Each dot represents a guide. Full representation was maintained after transduction with some correlation in abundance. Please click here to view a larger version of this figure.

Figure 2: Picture of the ultrasound-guided microinjection set-up. (A) Mouse carrying E9.5 embryos was anesthetized and placed on a heated platform with the uterus exposed in a PBS filled modified Petri dish chamber (stabilized by four modeling clay). The blue semi-round rubber supported the uterus containing embryos and the injection needle from the microinjector is positioned on the right side. The ultrasound scan head was mounted on the top relaying live image to the monitor behind with needle head visible. (B) Close-up ultra-sound image of an E9.5 embryo. Lentiviral library was injected with the needle (seen on right side) into the amniotic cavity. Please click here to view a larger version of this figure.
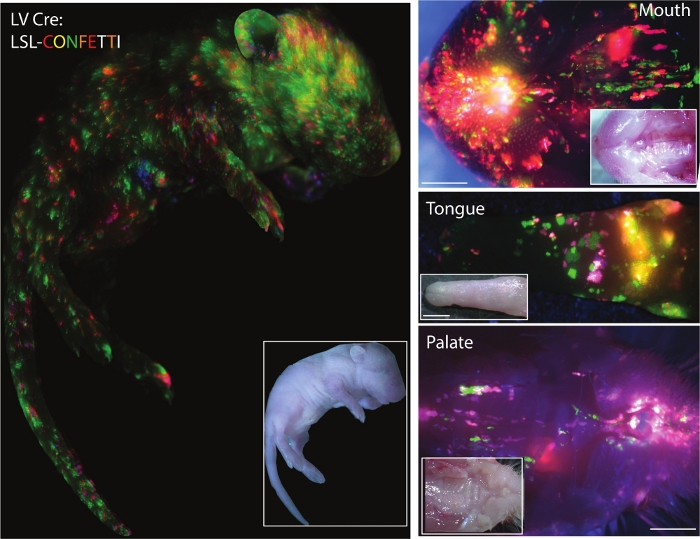
Figure 3: Successful transduction of surface epithelium in mouse. Fluorescent images of whole-body, oral cavity, tongue, and palate of newborn Cre-reporter LSL-Confetti mice transduced with Cre lentivirus (Inlet: corresponding white light images). Scale bars = 500 μm Please click here to view a larger version of this figure.
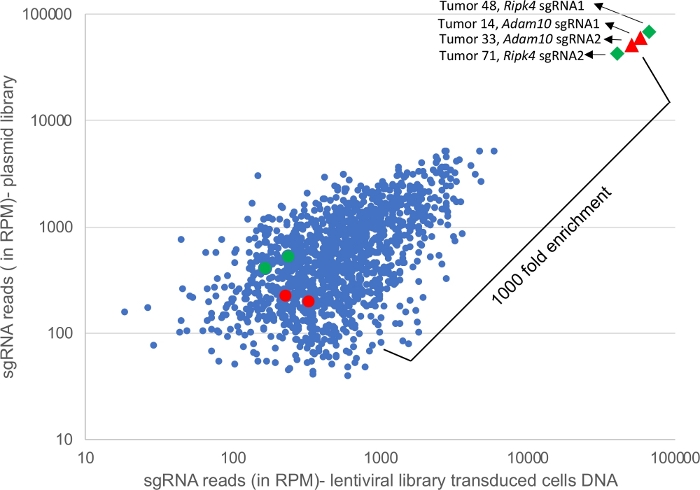
Figure 4: Deep sequencing of tumor samples. Graph showing sgRNA representation from a CRISPR library in plasmid DNA versus DNA from cells transduced with the same lentiviral library and in addition to reads from 4 representative tumors. The red and green circles denote the number of Adam10 and Ripk4 sgRNA reads in library and transduced cells whereas the red triangle represents the reads of Adam10 sgRNAs and the green diamond represents reads of Ripk4 sgRNAs identified in tumors from separate HNSCC mice showing a 1000x fold enrichment. Please click here to view a larger version of this figure.
| PCR1 Forward Primer | GAGGGCCTATTTCCCATGATTC |
| PCR1 Reverse Primer | CAAACCCAGGGCTGCCTTGGAA |
Table 1: Primers for PCR1 reaction. The forward and reverse primers used for amplification of the region surrounding sgRNA cassette from genomic DNA of cells transduced with the lenti virus.
| Step | Temperature | Time | |
| 1 | 98 °C | 30 sec | |
| 2 | 98 °C | 10 sec | |
| 3 | 66 °C | 30 sec | |
| 4 | 72 °C | 15 sec | 15 cycles (step 2-4) |
| 5 | 72 °C | 2 min | |
| 6 | 4 °C | Hold |
Table 2: PCR1 cycle parameters. PCR conditions used for the amplification of the region surrounding sgRNA cassette from genomic DNA of cells transduced with the lenti virus.
| 501 FW | AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACG ACGCTCTTCCGATCTtgtggaaaggacgaaaCACCG |
||
| 701 RV | CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGTGCT CTTCCGATCTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
||
| * The underlined bases indicate the Illumina (D501-510 for forward and D701-712 for reverse) barcode location that were used for multiplexing. | |||
| * Red colored bases indicate the sequence that bind to the target site on the lentiviral CRISPR plasmid. This region can be modified according to the lentiviral vector used. | |||
Table 3: Barcoding Primers for PCR2 reaction. Primers used to barcode amplify each tumor sample (either directly from genomic DNA or using the products from PCR1 as template). The underlined region indicates the unique barcode region that can be assigned for each sample for multiplexing in a deep sequencing reaction. The red colored base pairs indicate the target region in the lentiviral construct that these primers bind. This target region can be modified according to the type of lentiviral construct used.
| Step | Temperature | Time | |
| 1 | 98 °C | 30 sec | |
| 2 | 98 °C | 10 sec | |
| 3 | 68 °C | 30 sec | |
| 4 | 72 °C | 15 sec | 10 cycles (step 2-4) |
| 5 | 72 °C | 2 min | |
| 6 | 4 °C | Hold |
Table 4: PCR2 cycle parameters. PCR conditions used to barcode amplify each tumor sample (either directly from genomic DNA or using the products from PCR1 as template).
