Ion channels comprised of heteromers of leucine-rich repeat containing 8 (LRRC8) family proteins are found throughout vertebrate cells, participating in a wide range of physiological functions1,2. These LRRC8 channels, first identified as volume-regulated anion channels (VRACs) or volume-sensitive outwardly rectifying channels (VSOR), play a crucial role in cellular regulatory volume decrease3,4. They facilitate the expulsion of chloride ions and organic osmolytes, which is followed by water efflux in response to osmotic swelling. Beyond their role in osmotic stress response, their role in cellular volume regulation has been linked to cell proliferation and migration, apoptosis, spermiogenesis, and epithelial integrity5,6,7. Alteration of the membrane potential upon LRRC8/VRAC activation has been shown to contribute to myotube differentiation8 and insulin secretion by pancreatic β-cells9,10,11. Furthermore, LRRC8 channels conduct a variety of organic osmolytes such as purinergic signaling molecules ATP and cGAMP or the excitatory amino acid glutamate, placing these channels in cell-cell communication in the immune system or glia-neuron interaction12,13,14,15,16,17,18,19,20,21,22. Even xenobiotics, such as the dye fluorescein, the antibiotic blasticidin S or the anticancer drug cisplatin, are conducted by LRRC8 channels23,24,25.
There are numerous reports on the signal transduction leading to LRRC8/VRAC activation26,27,28. However, the mechanism remains unclear, and the literature presents a broad range of potential mechanisms that could depend on the specific physiological process. These include changes in cytosolic ion strength, interaction with the cytoskeleton, membrane composition, G proteins, the redox state, and phosphorylation cascades2,27,29,30,31.
LRRC8/VRAC channels contain LRRC8A as an essential subunit3,4 that must heteromerize with at least one of its paralogues LRRC8B-E to form physiologically functional channels4,14,32. The subunit composition determines biophysical properties of the channel, such as rectification and depolarization-dependent inactivation4,29,32,33,34, substrate specificity15,17,20,21,24,35, and some activation pathways36,37. Cryo-electron microscopy (cryo-EM) structures show that LRRC8A homomers, as well as heteromers, assemble as hexamers38,39,40, while LRRC8A/LRRC8C chimeras that form functional channels are heptamers41. The N-terminal part of all LRRC8 proteins comprises four transmembrane helices, and the C-terminal part contains a domain with leucine-rich repeats (LRRD). The available LRRC8 complex structures provide evidence that the LRRDs, which stretch into the cytosol3,4,23, may undergo conformational rearrangements during channel gating34,42,43. This notion is corroborated by the finding that C-terminal fusion of fluorescent proteins results in basal channel activity14 and that binding of nanobodies to the domains can modulate channel activity44. Moreover, conformational alterations of the C-termini were shown by intra-complex Förster resonance energy transfer (FRET)45.
The most common method to study ion channel activity is electrophysiological measurements46, which were extensively applied in the investigation of VRACs before their molecular identification47. However, there are various additional ways to monitor VRAC activity indirectly, including the measurement of its conducted substrates -halide ions or organic osmolytes- or its effect on cell volume48. In fact, the identification of LRRC8 proteins as VRAC relied on an assay based on the quenching of a halide-sensitive fluorescent protein49 by iodide entering the cell through activated VRACs3,4. Another method to monitor LRRC8/VRAC channel activity makes use of the movement of the cytosolic domains which can be observed, as in other ion channels50,51,52,53, by changes in FRET45. To this end, fluorescent proteins that serve as FRET pairs, such as cyan-fluorescent protein (CFP)/mCerulean3 as donor and yellow-fluorescent protein (YFP)/mVenus as acceptor, were fused to the C-termini of the LRRC8 proteins (Figure 1). Intra-complex FRET between LRRC8 subunits was shown by acceptor photobleaching experiments45. Avoiding the destructive photobleaching method, FRET changes over time were monitored by sensitized-emission FRET (SE-FRET), where basically the sensitized emission of the acceptor upon excitation of the donor due to the overlap of the emission spectrum of the donor with the excitation spectrum of the acceptor is measured. Application of extracellular hypotonicity, a stimulus for LRRC8/VRAC activation, resulted in a reversible reduction in SE-FRET intensity45. Importantly, simultaneous whole-cell patch-clamp measurements and FRET monitoring during hypotonic treatment showed that this reduction in FRET indeed mirrored LRRC8/VRAC activation45. This method, which avoids disrupting the plasma membrane or altering the intracellular environment by pipette solution, offers an alternative for monitoring LRRC8/VRAC activity. It is particularly useful in physiological settings where maintaining the native cytosol is crucial, subcellular resolution is necessary, or prolonged observation of channel activity is required.
Here, we present a protocol to study LRRC8/VRAC with such a FRET-based read-out. The protocol depicts how to handle and transfect cells, acquire sample and control images, analyze the data, and calculate sensitized emission FRET (SE-FRET) values.
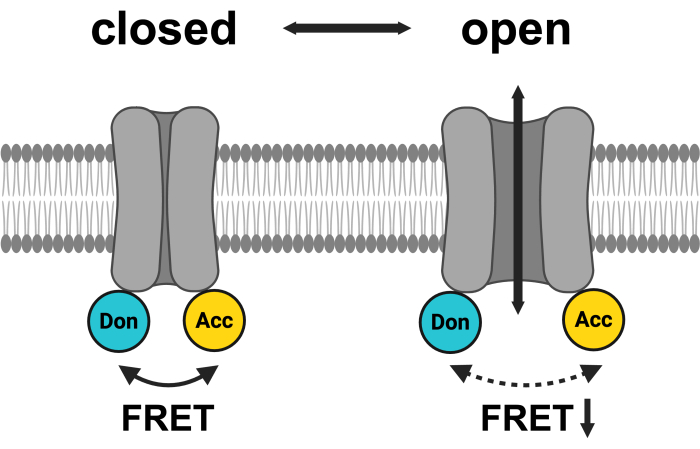
Figure 1: Schematic of the LRRC8 FRET pair system. mCerulean3 is shown in cyan, and mVenus is shown in yellow. Following VRAC opening, the distance (and/or the spatial orientation) between the fluorophores changes, resulting in a reduced energy transfer between donor (Don) and acceptor (Acc) and, in turn, lowering the observed FRET. Created with BioRender.com. Please click here to view a larger version of this figure.
1. Preparation of buffer and reagents
- Prepare medium and isotonic, hypotonic, and hypertonic buffers and measure the osmolarity of the buffers with an osmometer to ensure that the osmolarity (Osm) is in the expected range with only an acceptable deviation.
- Cell culture media: Prepare Dulbecco´s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin
- Isotonic imaging buffer (~340 mOsm): Prepare the isotonic imaging buffer by mixing 150 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM D(+)-Glucose and 10 mM 4-(2-Hydroxy Ethyl)-1-Piperazine Ethane Sulfonic acid (HEPES).
- Hypotonic imaging buffer (~250 mOsm): Prepare the hypotonic imaging buffer by mixing 105 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM D(+)-Glucose and 10 mM HEPES.
- Hypertonic imaging buffer (~500 mOsm): Prepare the hypertonic imaging buffer by mixing 160 mM D(-)-Mannitol, 150 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM D(+)-Glucose and 10 mM HEPES.
NOTE: Buffers may be supplemented with specific drugs depending on what will be investigated.
2. Growth of adherent cells on glass bottom dishes
- Prepare at least one dish for transfection only with the protein of interest (POI) fused to either the donor or the acceptor fluorophore and at least one dish for transfection with the donor and acceptor pair (Table 1 and Figure 2). Use glass-bottom dishes to allow for fluorescence microscopy. Depending on the cell type, pretreat the surface by special cleaning or coating it with polylysine.
NOTE: Here, FRET donor mCerulean3 (mCer)54 and FRET acceptor mVenus (mVen)55 are used as FRET pair, but other combinations of fluorophores are also applicable. Sample expressing the POI fused to the donor or acceptor alone are used to determine the correction factors β and γ (Table 1) necessary to correct for donor bleed through in the acceptor channel and cross excitation of the acceptor by the donor excitation wavelength (Figure 2Ac, Bf). - The day before transfection, seed 1 x 105 HeLa cells in 2 mL of cell culture media on 35-mm dishes with a glass bottom suitable for fluorescence microscopy. Culture cells overnight at 37 °C and 5% CO2 atmosphere in a cell culture incubator.
NOTE: Volumes of reagents can be adapted to the surface area of other cell culture dishes/flask with a different size. The cell number can be adapted to a density suitable for different cell lines and experimental approaches.- Aspirate the cell culture supernatant from the monolayer of adherent cells cultured on T75 cell culture flasks using a vacuum pump.
- Wash the cells by adding 10 mL of Dulbecco's Phosphate Buffered Saline (DPBS) to the cell culture flask and aspirating the solution afterward with a vacuum pump.
- Cover the cells completely with 1mL of 0,05% Trypsin-EDTA and incubate the cells for 2 min at 37 °C and 5% CO2 atmosphere in a cell culture incubator.
- Add 9 mL of cell culture media and suspend the cells by pipetting.
- Transfer the cell suspension into a 15 mL tube and centrifuge at 700 x g for 3 min at room temperature (RT).
- Aspirate the supernatant with a vacuum pump and resuspend the cell pellet in 10 mL of cell culture media.
- Mix equal volumes of cell suspension and 0.4% trypan blue in a tube and add 10 µL to a counting slide. Place the slide into the automated cell counter and start counting using the appropriate program for cell counting.
- To calculate the volume of cell suspension needed for each dish, divide the cell number needed per dish (1 x 105) by the cell number per milliliter obtained from the cell counter.
- Prepare a cell suspension in a suitable tube containing the number of cells needed per dish in the volume needed per dish (2 mL) plus the amount for one dish extra.
- Mix cell suspension at least 20 times by inverting the tube and pipette 2 mL of the cell suspension in each dish.
- To ensure a more even cell distribution, leave the dishes for 30 min at RT before placing them in the cell culture incubator.
3. Cell transfection
NOTE: Here, FuGENE was used as a transfection reagent. Other transfection reagents and methods are also applicable. Optimal ratios of plasmid DNA (pDNA) to transfection reagents and time for optimal expression for each POI and cell model need to be assessed empirically. Here, 2 µg of total pDNA per 35 mm dish was used. FRET donor and acceptor constructs were used in a 1:1 ratio, and the pDNA-to-transfection reagent ratio was 1:4 (Table 1).
- Before transfection, replace the cell culture supernatant with fresh 2 mL of pre-warmed media.
- Dilute pDNA in Opti-Minimal Essential Medium (MEM) with a final volume of 100 µL (=pDNA solution).
- Dilute the transfection reagent in Opti-MEM with a final volume of 100 µL (=reagent solution).
NOTE: For a more uniform transfection, prepare a master mix for the pDNA and reagent solution. - Mix pDNA and reagent solution well.
- Add the pDNA solution into the reagent solution (=transfection solution).
- Mix the transfection solution well.
- Incubate the transfection solution for 15 min at RT.
- Add the transfection solution dropwise in a spiral motion to the dish.
- Mix by moving the dish 5 times horizontally and vertically on the surface of the bench.
- Culture cells overnight at 37 °C and 5% CO2 atmosphere in a cell culture incubator.
| Condition | Construct (s) | Sample | Used for |
| 1 | LRRC8A-mCerulean | Donor construct only | determine correction factor β |
| 2 | LRRC8E-mVenus | Acceptor construct only | determine correction factor γ |
| 3 | LRRC8A-mCerulean and LRRC8E-mVenus | FRET pair | SE-FRET quantification |
Table 1: Example of conditions necessary for a typical SE-FRET experiment to measure LRRC8/VRAC activity of a channel composed of the LRRC8A subunit fused to the donor (mCerulean3) and LRRC8E subunit fused to the acceptor (mVenus) fluorophore.
4. Image acquisition for correction factor determination
NOTE: There is a bleed-through of the donor emission into the detected emission of the acceptor during FRET. Moreover, there is cross-excitation of the acceptor fluorophore by the donor excitation wavelength. These processes must be compensated for during the calculation of the SE-FRET. To this end, correction factors are determined in cells expressing only the FRET donor or acceptor 24 h after transfection. Here, the imaging was carried out on a Leica THUNDER Imager equipped with a Leica LED8 lamp, the filter cube CYR71010, an HC PL APO 63x/1.40 OIL objective, long pass filter for 460/80 and 553/70 and a Leica DFC9000GTC camera. Experiments were performed without environmental control but in the presence of HEPES in the imaging buffers to stabilize the pH. For long-term observation/measurements, it is advised to use an environmental control system. For analysis, SE-FRET is calculated from the captured raw images. This can be done simultaneously during acquisition or afterward. Here, the Leica LAS X software with the SE-FRET plugin was used to simplify the experimental procedure regarding the calculation of the correction factors and visualizing the SE-FRET value changes in real-time during image acquisition. For post-acquisition, correction factors and SE-FRET can be determined with other software packages (e.g., FIJI) after raw data acquisition according to the protocol provided below.
- Before image acquisition of single transfected cells to calculate the correction factors, use one sample expressing the FRET pair to set up the microscopy setting for all channels (donor excitation/donor emission DD, donor/acceptor DA, and acceptor/acceptor AA; see Table 1 and Figure 2C and Figure 3A).
NOTE: For SE-FRET measurements with mCerulean3- and mVenus-tagged VRAC subunits transfected as described above, the following parameters were used: 8×8 pixel binning, 100ms exposure time, and 10% LED lamp intensity. - Take the sample expressing the donor construct only (Figure 2A), aspirate the cell culture media, and wash the cells three times with 2 mL of isotonic buffer.
- Add 3 mL of isotonic buffer and place the sample on the microscope stage.
- Find a field of view (FOV) with at least one cell expressing the donor construct.
- Image all the channels (DD, DA, and AA see Table 2 and Figure 2A).
- Draw a region of interest (ROI) around the cell/cells and measure the mean intensity of DA (= IDA; Figure 2Ac) and DD (= IDD; Figure 2Aa).
- For background subtraction, draw an ROI in the DA and DD channels where only the background signal is found and measure the mean intensity (IBDA and IBDD).
- Subtract the mean intensity of the background ROI from the mean intensity measured in the cell ROI of the corresponding channel (
 and
and  ).
). - Repeat steps 4.2- 4.5 with the sample expressing only the acceptor construct (Figure 2B).
- Draw an ROI around the cell(s) and measure the mean intensity of DA (= IDA; Figure 2Bf) and AA (= IAA; Figure 2Be).
- For background subtraction, draw an ROI in the DA and AA channels where only the background signal is found and measure the mean intensities (IBDA and IBAA).
- Subtract the mean intensity of the background ROI from the mean intensity measured in the cell ROI of the corresponding channel ( and
 ).
). - Use the values determined for IDA*, IDD*, and IAA* to calculate the correction factors
 and
and  ,
,
Whereby the correction factor β is used to compensate for the bleed-through of the donor emission into the DA channel. The correction factor γ is used to compensate for the cross-excitation of the acceptor fluorophore by the donor excitation wavelength in the DA channel.
| Excitation of | Emission of | Channel name | LED line | Filter cube | Long-pass filter |
| Donor | Donor | DD | 440 nm | CYR71010 | 460/80 nm |
| Donor | Acceptor | DA | 440 nm | CYR71010 | 535/70 nm |
| Acceptor | Acceptor | AA | 510 nm | CYR71010 | 535/70 nm |
Table 2: Summary of channels required for SE-FRET experiments.

Figure 2: Representative fluorescence images of samples used to calculate the correction factors β and γ necessary to determine the VRAC activity of a channel composed of LRRC8A subunit fused to the donor mCerulean3 (mCer) and LRRC8E subunit fused to the acceptor mVenus (mVen) fluorophore by SE-FRET measurements. (A,B) Detection of the donor/donor DD, acceptor/acceptor AA, and donor/acceptor DA channel in HeLa cells only expressing the donor (A) LRRC8A-mCer or the acceptor (B) LRRC8E-mVen. (C) Detection of DD, AA, and DA channels in HeLa cells co-transfected with the donor and acceptor pair LRRC8A-mCer and LRRC8E-mVen. Panels a-i show images taken in the donor detection channel (excitation of the donor and detection of the donor signal; DD; a, d and g), the acceptor detection channel (excitation of the acceptor and detection of the acceptor signal; AA; b, e, and h ) and the FRET signal detection channel (excitation of the donor and detection of the acceptor signal; DA; c, f, and i). Panel j is the overlay of panels g and h. DD channel is shown in green and AA channel in magenta. Scale bar = 10 µm. Please click here to view a larger version of this figure.
5. Time-lapse imaging for SE-FRET quantification
- Take the sample expressing the donor and acceptor construct (Table 1, Figure 2C and Figure 3A), aspirate cell culture media, and wash the cells three times with 2 mL of isotonic buffer 24 h after transfection.
- Add 3 mL of isotonic buffer and place the sample on the microscopy stage.
- For later aspiration of the isotonic buffer, fixate and adjust a hose cannula so the cannula tip reaches the bottom of the dish.
- For adding buffers, fixate and adjust tubings so that the buffer driven by gravity flow can drop into the dish.
NOTE: Here, a valve controlled gravity perfusion system with four magnetic valves to control the buffer flow was used, but other methods are also applicable. - Find a FOV with at least one cell expressing the donor and acceptor construct simultaneously.
- Set up a time-lapse experiment for the channels DD, DA, and AA with an interval of 10 s and a duration to cover all conditions of the stimulation sequence, e.g., 12 cycles of baseline followed by 15 cycles per condition (Figure 3).
NOTE: The interval and the number of cycles can be adopted according to the experimental needs but should always include a baseline measurement for normalization for better visualization and comparison of the data. - After baseline measurement, wash the sample in the buffer for the first condition.
- Aspirate the isotonic buffer via the hose cannula, applying a vacuum with a syringe.
- Add 3 mL of the buffer of the next condition by gravity flow.
- Aspirate the buffer and add again 3 mL of the same buffer.
- Repeat step 5.7.3 once more.
- After measurement of the first condition, wash the sample in the buffer for the next condition (steps 5.7.1-5.7.4).
- Repeat steps 5.5-5.8 until all conditions are captured.
- For SE-FRET quantification, draw an ROI around the cell/cells and measure the mean intensity in the DD = (IDD), DA (IDA), and AA (IAA) channel for all images (time points) in the time series (Figure 3A, B).
- For background subtraction for the FRET signal, draw an ROI in the DA channel where only the background signal is found and measure the mean intensity (IBDA).
- Subtract the mean intensity of the background ROI from the mean intensity measured in the cell ROI of the DA channel ().
- Use the values determined to calculate the mean SE-FRET values for each ROI and all time points
 .
. - For better comparability of different conditions and visualization of the SE-FRET values, use the mean of the baseline values of each ROI to normalize all time points of the corresponding ROI.
- Plot the normalized SE-FRET values over time (Figure 3B).

Figure 3: Representative fluorescence images and SE-FRET quantification. (A) Representative fluorescence images and the apparent SE-FRET of the first time point of a time laps experiment to measure the VRAC activity by SE-FRET of a channel composed of LRRC8A and LRRC8E subunits depending on the tonicity. Scale bar = 10 µm. The same cells are shown in Figure 2C. Panels a–i show the detection of the DD, AA, and DA channels and the calculated apparent SE-FRET. White outlines represent the ROIs (cells i-iii in panel d) used to measure the mean signal intensities in DD, AA and DA and the apparent SE-FRET image. (B) Quantification of the SE-FRET values over time. Sequence of conditions was 12 cycles of isotonic imaging buffer (baseline) followed by 15 cycles of hypotonic and 15 cycles of hypertonic imaging buffer. The raw mean SE-FRET value of each ROI (cells i-iii) and time point was normalized to the mean of the baseline (isotonic) value for the corresponding ROI. Please click here to view a larger version of this figure.
With this FRET-based method, LRRC8/VRAC activity can be monitored during osmotic stimulation, and the reduction in SE-FRET correlates with the degree of extracellular hypotonicity45. Representative results for hypotonicity-induced channel activation are also shown here (Figure 3 and Figure 4). In addition, LRRC8/VRAC activation by different isosmotic stimuli, such as manipulation of diacylglycerol signaling45 or during myocyte activation56, can be observed.

Figure 4: SE-FRET traces. (A) Apparent SE-FRET traces from 5 independent experiments. Data represent mean ± SD of N = 2 to N = 7 cells per field of view (FOV). (B) Mean ± SD of all cells (N = 31 cells). Please click here to view a larger version of this figure.
As LRRC8/VRAC channels are also involved in apoptotic volume decrease24,57, observing channel activity upon induction of apoptosis would be another application for the method described here. Accordingly, the protocol to monitor SE-FRET in HeLa cells expressing LRRC8A-mCerulean3 and LRRC8E-mVenus was carried out while applying death receptor-mediated apoptosis-inducing drugs. Tumor necrosis factor (TNF)-α and cycloheximide (CHX) were previously shown to evoke VRAC currents within a few minutes58. After the addition of 2 ng/mL TNF-α and 1 µg/mL CHX in isotonic buffer, there was a robust decrease in SE-FRET (Figure 5). Replacing the buffer by a hypertonic medium, even though containing TNF-α and CHX, SE-FRET values recovered close to baseline (Figure 5A), corresponding to diminishing VRAC currents in hypertonic bath solution during treatment with apoptosis-inducing Fas ligand58. Treatment of the cells with DMSO, the solvent for TNF-α and CHX, did not result in SE-FRET reduction. TNF-α + CHX did not affect SE-FRET of CFP-18aa-YFP, an EYFP and ECFP tandem construct as FRET control59, demonstrating the specificity for LRRC8/VRAC (Figure 5B).
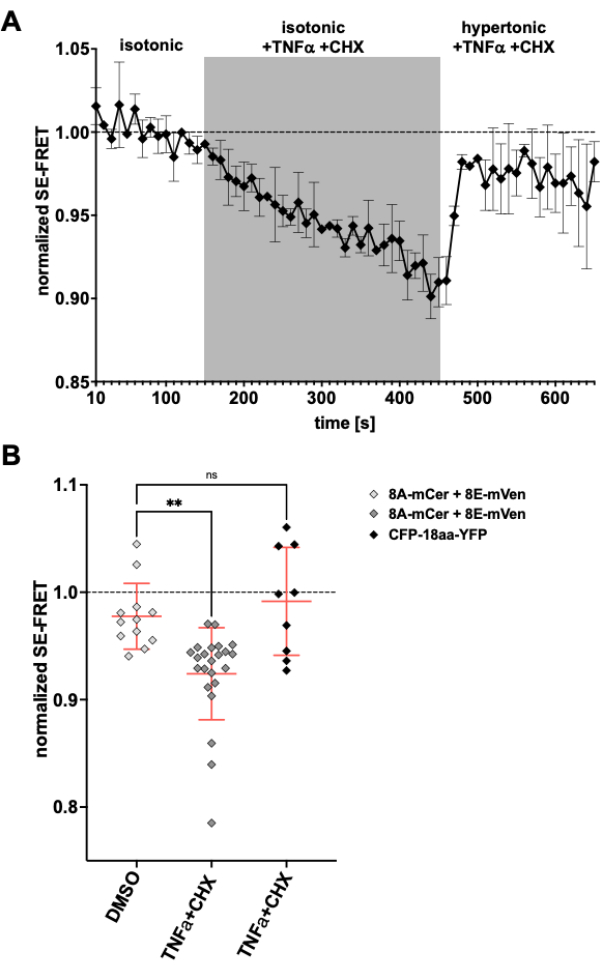
Figure 5: Isosmotic VRAC activation by death receptor-mediated apoptosis. (A) Normalized SE-FRET values from LRRC8A-mCer/LRRC8E-mVen-expressing HeLa cells (n = 8 dishes, 23 cells) over time. After 15 cycles in isotonic imaging buffer (baseline), the bath solution was replaced by isotonic buffer supplemented with 2 ng/mL TNF-α and 1 µg/mL cycloheximide (CHX) for 30 cycles followed by 20 cycles hypertonic imaging buffer with TNF-α and CHX. The raw mean SE-FRET value of each ROI and time point was normalized to the mean of the baseline (isotonic) value for the respective ROI. (B) Quantification of normalized SE-FRET values of LRRC8A/E-expressing HeLa cells as in A with isotonic solution containing DMSO as vehicle control for CHX (n = 5 dishes, 12 cells) or isotonic buffer containing apoptosis inducers as in A (n = 8 dishes, 23 cells), or of HeLa cells expressing CFP-18aa-YFP with apoptosis inducers (n = 3 dishes, 9 cells). Data represent the mean of the last 10 time points in the respective buffer of individual cells (symbols) and mean of all cells ± SD; ** p < 0.01 ordinary one-way ANOVA followed by Tukey´s multiple comparison post-hoc test. Please click here to view a larger version of this figure.
