Isolation of Active Caenorhabditis elegans Nuclear Extract and Reconstitution for In Vitro Transcription
Summary
Here, we describe a detailed protocol for isolating active nuclear extract from larval stage 4 C. elegans and visualizing transcription activity in an in vitro system.
Abstract
Caenorhabditis elegans has been an important model system for biological research since it was introduced in 1963. However, C. elegans has not been fully utilized in the biochemical study of biological reactions using its nuclear extracts such as in vitro transcription and DNA replication. A significant hurdle for using C. elegans in biochemical studies is disrupting the nematode's thick outer cuticle without sacrificing the activity of the nuclear extract. While several methods are used to break the cuticle, such as Dounce homogenization or sonication, they often lead to protein instability. There are no established protocols for isolating active nuclear proteins from larva or adult C. elegans for in vitro reactions. Here, the protocol describes in detail the homogenization of larval stage 4 C. elegans using a Balch homogenizer. The Balch homogenizer uses pressure to slowly force the animals through a narrow gap breaking the cuticle in the process. The uniform design and precise machining of the Balch homogenizer allow for consistent grinding of animals between experiments. Fractionating the homogenate obtained from the Balch homogenizer yields functionally active nuclear extract that can be used in an in vitro method for assaying transcription activity of C. elegans.
Introduction
The small, free-living nematode Caenorhabditis elegans is a simple yet powerful model organism for addressing a wide range of biological questions. Since its introduction in 1963, the nematodes have been invaluable to answering questions in neurobiology, metabolism, aging, development, immunity, and genetics1. Some of the animal's many characteristics that make it an ideal model organism include short generation time, the effectiveness of RNA interference, transparent body, and the completed maps of both its cellular lineage and nervous system.
While the nematode's contributions to science are vast, they have been under-utilized to elucidate the eukaryotic transcription system, with most of our understanding about these mechanisms coming from studies using nuclear extract from yeast, fruit fly, and mammalian cell culture2. The biggest hurdle that dissuades researchers from extracting functional nuclear extract is the nematode's tough outer cuticle. This exoskeleton comprises cross-linked collagens, cuticlins, glycoproteins, and lipids, making C. elegans from larval stage to adulthood resistant to protein extraction via chemical or mechanical forces3. An in vitro transcription system using C. elegans nuclear extract was once developed but not widely adopted due to the system's limited scope, and the use of Dounce homogenizer for preparing the extract could lead to protein instability4,5.
Unlike the previous protocol for nuclear extract isolation which utilized a Dounce homogenizer to break C. elegans, this protocol uses a Balch homogenizer. The Balch homogenizer consists of two main components: a tungsten carbide ball and a stainless-steel block with a channel bored through from one end to another. The Balch homogenizer is loaded with the tungsten carbide ball and capped on either side to seal the grinding chamber. Syringes can be loaded on the two vertical ports leading into the grinding chamber. As the material is passed from one syringe to the other through the grinding chamber, the pressure from the syringes forces the material through a narrow gap between the ball and the wall of the chamber. This slow and constant pressure breaks the material until it reaches a consistent size that is able to pass through the narrow gap easily. Forcing C. elegans through the narrow gap via a constant yet gentle pressure breaks the animals open, releasing their content into the surrounding buffer. Switching ball sizes further tightens the gap, breaking the newly released cells and frees the nuclei into the buffer. Multiple instances of centrifugation separate the nuclei from the rest of the cell debris, allowing for the collection of a clean nuclear extract. The Balch homogenizer is preferred over the Dounce homogenizer for several reasons: the system can handle a large number of animals, making it possible to extract a high amount of active proteins in a single attempt; the precise machining of the balls and the steel block allows for consistent grinding between multiple samples; the heavy steel block act as a heat sink, uniformly drawing heat away from the grinding chamber, preventing denaturing.
After isolation, the nuclear extract transcriptional activity must be verified before being used in any biochemical experiments. Traditionally, transcription activity was measured using radiolabeled nucleotides to track and visualize the newly synthesized RNA. However, radioactive labeling can be burdensome as it requires precaution during use and disposal6. Technological advancements allow researchers today to use much less harmful or troublesome methods to measure even small RNA quantities using techniques such as quantitative real-time PCR (qRT-PCR)7. Here, the protocol describes a method to isolate active nuclear extract from larval stage 4 (L4) C. elegans and visualize transcription activity in an in vitro system.
Protocol
1. Media preparations
- Prepare sterile lysogeny broth (LB) agar plates and liquid media following the manufacturer's instructions.
- Streak Escherichia coli (E. coli) strain OP50 on a LB agar plate. Incubate the bacteria streak at 37 °C overnight.
- Store the E. coli OP50 streak plate at 4 °C after incubation. The E. coli OP50 plate can safely be stored at 4 °C for 2 weeks if wrapped in parafilm to prevent moisture loss.
- Prepare 2 L of Nematode Growth Media (NGM) using the recipe in Table 1.
NOTE: Nystatin is optional. Nystatin helps prevent mold, and other fungal contaminates from growing on the NGM plates. The large 150 mm diameter plates have a higher chance of catching fungal spores when the lid is removed for pouring and seeding E. coli OP50. Nystatin can be purchased premixed in sterile solution from vendors at 10,000 units/mL or can be purchased as a sterile powder and mixed with sterile water. Nystatin cannot be autoclaved, nor can it be effectively filter sterilized. Attention to the aseptic technique is crucial while mixing a solution of nystatin in the lab. Autoclave 1 M CaCl2, 1 M KPO4 pH 6.0, and 1 M MgSO4 at 121 °C for 15 min. Filter sterilize cholesterol through a 0.22 µm filter after being dissolved in 95% ethanol. - After adding the reagents to the media, pour NGM into forty 150 mm Petri dishes. Each 150 mm dish requires 50 mL to fill. Let the NGM plates cool overnight at room temperature.
- Inoculate two 50 mL conical tubes containing 25 mL of sterile LB with E. coli OP50 from the previous streak plate. Incubate the broth at 37 °C for 24 h in a shaking incubator maintained at 200 rpm.
NOTE: For consistency, inoculate both tubes with the same single colony from the streak plate. If this proves difficult, inoculate 5 mL of sterile LB with a single colony in a 50 mL conical tube and incubate the culture for 16 h at 37 °C in a shaking incubator maintained at 200 rpm the day before preparing the NGM plates. Store the fresh liquid culture at 4 °C for up to two days. Inoculate the two 25 mL of broth with 25 µL of liquid culture and incubate under the same conditions as mentioned above. - Seed fresh NGM plates with 1 mL of fresh E. coli OP50 liquid culture and spread with a flame sterilized spreader evenly across the surface of the plate aseptically to create a large bacteria lawn that covers the majority of the plate, taking care not to spread the bacteria from edge to edge. Allow the E. coli OP50 to grow at room temperature for 72-96 h or until a visibly thick lawn appears.
NOTE: After 24 h, move the freshly seeded NGM plates to a covered container to reduce moisture loss and extend the life of the plates.
2. Animal preparations and bleach synchronization
- Transfer 5 well-fed, wild-type, gravid adult C. elegans to each of the 10 fresh NGM plates seeded with E. coli OP50, for a total of 50 animals.
- Allow the animals to lay eggs. Let the progeny grow at 20 °C until they reach the gravid adult stage.
- Use 15 mL of M9 buffer (Table 2) to collect the new well-fed wild-type, gravid adults from the ten maintenance plates and transfer the animals to a labeled 15 mL conical tube.
- Centrifuge the animals at 1,000 x g for 3 min to pellet all animals at the bottom of the tube.
- In a separate, labeled 15 mL conical tube, mix 2 mL of bleach with 5 mL of 1 N NaOH for bleach synchronization. Vortex the solution to mix thoroughly.
NOTE: Use the bleach + NaOH solution the same day it is prepared to be effective. - Gently remove the supernatant from the centrifuged animals using a 10 mL sterile pipette. Attempt to remove as much M9 buffer as possible to improve the breaking of animals.
- Add 500 µL of the bleach + NaOH solution to the animal pellet and start a timer for 4 min.
- Either by hand or using a rocker, gently rock the tube to break the animal pellet completely and let the animals move freely in the bleach + NaOH solution. Continue rocking the tube for the whole 4 min duration.
NOTE: The amount of bleach + NaOH solution needed can vary depending on the size of the animal pellet being synchronized. Optimize this technique beforehand with varying amounts of animals and bleach + NaOH solution. - After 4 min, check the breaking efficiency under a dissection microscope. Ensure that a majority of the gravid adult animals are broken, and their internal contents released, including the eggs.
- If the animals are not split open, briefly vortex the tube at maximum speed, then check again under the microscope.
NOTE: While the eggs are resistant to the bleach + NaOH solution, they are not impervious. Eggs must not be exposed to the bleach + NaOH solution any longer than necessary. Stalling for too long during the animal breaking step can lead to developmental problems for the progeny. - Add 10 mL of the M9 buffer to the broken animal solution.
- Centrifuge the eggs and the remains at 1,000 x g for 3 min.
- Pipette the supernatant away, making sure not to touch the new pellet containing all the eggs at the bottom of the tube.
- Add another 10 mL of the M9 buffer and centrifuge again for 3 min at 1,000 x g.
- Repeat steps 2.13-2.14 two more times to ensure that none of the bleach + NaOH solution remains.
- After the third M9 buffer wash, remove the supernatant and add 10 mL of the S-basal buffer (Table 2).
- Invert the tube to break the pellet at the bottom to suspend the eggs equally in the buffer.
- Place the tube on a rocker and gently rock the tube for 22 h at 20 °C to allow the eggs to hatch and reach larval stage 1 (L1) arrest.
- After 22 h, centrifuge the tube containing synchronized L1 animals for 3 min at 1000 x g.
- Pipette and discard the supernatant leaving approximately 1 mL of buffer in the tube.
- Using a micropipette, disturb the animal pellet to make a homogeneous suspension of L1 animals in the remaining buffer.
- Transfer a single droplet of the homogeneous suspension of L1 animals to a labeled 150 mm NGM plate seeded with E. coli OP50.
- Calculate the animal density per drop by visually counting the number of L1 animals per drop using a dissection microscope. A 150 mm NGM plate with a fully grown lawn of E. coli OP50 can support up to 500 animals of the L1 stage to reach the gravid adult stage.
- Transfer the L1 animals to six 150 mm NGM plates with E. coli OP50. Ensure not to overload the plate.
NOTE: If it is unclear whether the animals will have enough food for development between L1 and gravid adult, add fewer animals to each plate and use more plates. - Allow the animals to grow for 72 h at 20 °C until they reach the gravid adult stage and begin laying eggs.
- Perform a second round of bleach synchronization on the synchronized, well-fed wild-type gravid adult animals.
- Place the synchronized L1 animals on ten 150 mm NGM plates seeded with E. coli OP50, with approximately 1,000 animals per plate.
- Allow the new synchronized L1 animals to grow for 48 h at 20 °C until they reach the L4 stage.
NOTE: The goal is to obtain an approximately 700 – 800 µL pellet of L4 animals. Too many animals can make it difficult to use the Balch homogenizer; this can lead to muscle strain while operating the homogenizer and possible leakage from the syringes due to the increased pressure.
3. Balch homogenizer preparation
- Prepare both hypotonic and hypertonic buffers as mentioned in Table 3 and Table 4.
NOTE: The hypotonic and hypertonic buffers can be prepared in advance and stored safely at 4 °C. - Clean the Balch homogenizer by flooding the grinding chamber with 70% ethanol, then rinse the chamber with deionized water to remove excess ethanol.
NOTE: Avoid using any caustic agents to clean the Balch homogenizer. Thorough rinsing with ethanol and deionized water should be sufficient to clean it. - Insert the 7.9820 mm (18 µm gap clearance) tungsten carbide ball into the grinding chamber.
- Cap each end of the barrel of the Balch homogenizer and secure the caps with the provided thumbscrews.
- Prepare 5 mL of 'complete hypotonic buffer' per sample: Add 5 µL of 1 M DTT (final concentration: 1 mM DTT) and 100 µL of 100x protease inhibitor, single-use cocktail (final concentration: 2x). Keep the buffer on ice.
- Prepare 5 mL of 'complete hypertonic buffer' per sample: Add 5 µL of 1 M DTT (final concentration: 1 mM DTT) and 100 µL of 100x protease inhibitor, single-use cocktail (final concentration: 2x). Keep the buffer on ice.
NOTE: Use the complete hypotonic and hypertonic buffer the same day of preparation. Do not store it for later use. Store 1 M DTT as single-use aliquots at -20 °C. Store the protease inhibitor single-use cocktail at 4 °C. - Fill a sterile 2 mL syringe with 1 mL of 'complete hypotonic buffer' and gently flush the grinding chamber of the Balch homogenizer. Leave approximately 500 µL of 'complete hypotonic buffer' in the chamber.
- Store the flushed homogenizer on ice and let it cool for 30 min.
NOTE: The homogenizer must be ice cold before grinding the animals. The grinding process can produce heat due to friction and denature the nuclear proteins. Ensure that the metal homogenizer is ice cold to help prevent this from happening. Be sure to avoid any water from the surrounding ice from entering the homogenizer. Use two sterile syringes to plug the holes in the homogenizer and block any unintentional liquids from entering the grinding chamber.
4. Collection of animals
NOTE: A quick reference guide is provided, marking the major steps for collection, disruption, and fractionation of the animals (Figure 1).
- Collect the well-fed L4 animals with M9 buffer in a 15 mL conical tube and centrifuge the animals at 1000 x g for 3 min. Remove the supernatant and continue washing the animal pellet until the supernatant is clear.
- Wash the animals with 3 mL of 4 °C hypotonic buffer and centrifuge again at 1000 x g for 3 min.
NOTE: During the final wash with the 4 °C hypotonic buffer, the animals may stick to the side of the tube. This is normal and may result in a small loss of animals during the removal of the supernatant. - Remove the hypotonic buffer and add 1 mL of "complete hypotonic buffer" to the animal pellet. Transfer the animal suspension to a new 2 mL sterile syringe.
NOTE: While transferring the animals to the syringe using a micropipette, gently pipette a sterile 0.1% Tween20 solution to coat the inside of the pipette tip to reduce the number of animals lost due to sticking to the pipette tip walls.
5. Fractionation
- On ice, homogenize the animals by gently pushing the animals through the grinding chamber of the Balch homogenizer loaded with the 7.9820 mm ball and into a new sterile syringe. Repeat pushing the animals through the grinding chamber for 30 complete cycles.
NOTE: A 'complete cycle' is defined as the complete up and down motion of a syringe's plunger.
This grinding step uses 7.9820 mm ball (18 µm ball bearing). - After 30 cycles, remove as much of the animal suspension as possible from the Balch homogenizer and store the syringe, tip down in a 1.7 mL microtube.
- Remove the 7.9820 mm ball from the grinding chamber and clean it with deionized water. Dry and return the ball to its labeled tube.
- Insert the 7.9880 mm (12 µm gap clearance) ball into the grinding chamber and reseal the homogenizer.
- Flush the grinding chamber again with 1 mL of ice-cold 'complete hypotonic buffer'.
- Grind the suspension for 25 complete cycles.
- After 25 cycles, remove the animal suspension from the Balch homogenizer, transfer the suspension into a clean 1.7 mL microtube, and store it on ice.
NOTE: Disassemble and clean the Balch homogenizer with 70% ethanol and deionized water. Be sure to return the 7.9880 mm ball to its proper tube. - Pellet the animal bodies and debris by centrifuging the suspension at 500 x g, 4 °C for 5 min.
- Pipette 40 µL of the supernatant to a tube labeled 'input fraction' and store it on ice.
NOTE: Write all labels with an alcohol-proof pen to avoid smearing them later. - Transfer the remaining supernatant to a new 1.7 mL tube, taking care to avoid touching the pellet at the bottom of the tube and then discard the pellet.
- Centrifuge the supernatant to pellet the nuclei at 4,000 x g, 4 °C for 5 min.
- Transfer the supernatant, careful not to disturb the pelleted nuclei, to a new 1.7 mL tube and label the tube as 'cytosolic fraction'.
NOTE: Centrifuge the cytosolic fraction further at 17,000 x g, 4 °C for 30 min to remove any remaining insoluble material, and use it as a negative control for western blots (specifically for nuclear proteins). - Wash the nuclei pellet with 500 µL of 'complete hypotonic buffer' and transfer the pellet to a new 1.7 mL tube. Centrifuge the suspended pellet at 4,000 x g, 4 °C for 5 min.
- Discard the supernatant and add 500 µL of fresh 'complete hypotonic buffer' to the nuclear pellet and transfer the suspension to a new 1.7 mL tube. Centrifuge the sample again at 4,000 x g, 4 °C for 5 min.
- Remove the supernatant and dissolve the pellet in 40 µL of 'complete hypertonic buffer'. Transfer the new nuclear suspension to a new 1.7 mL tube, label the tube as 'nuclear fraction', and store it on ice.
- Determine the protein concentration of the three fractions using a fluorescent quantification kit.
NOTE: The amount of nuclear protein acquired from this method can range from 1-2 µg/µL. - Aliquot the nuclear fractions into single-use tubes containing 6 µg of the nuclear protein and snap freeze in a dry ice and ethanol bath. Store at -80 °C until further use.
6. Transcription assay
- Turn on and preheat a heat block to 30 °C.
- Remove the following from the Nulcear Extract in vitro transcription system: 50 mM MgCl2, Nuclear Extract 1x Transcription Buffer, 100 mM rATP, 100 mM rCTP, 100 mM rGTP, 100 mM rUTP, and the CMV promoter positive-control template. Thaw on ice.
NOTE: Amplify the positive-control DNA template using a high-fidelity polymerase and store it as normalized single-use aliquots. - Mix and centrifuge the thawed rNTPs before preparing a 10 mM working solution.
NOTE: Add 2 µL of each rATP, rCTP, rGTP, and rUTP to 12 µL of H2O to achieve 10 mM of each rNTP. This composition can be scaled up if necessary. - Aliquot the rNTP mixture to labeled single-use tubes and store at -20 °C for future use.
- In a fresh 1.5 mL tube labeled "Mastermix" add the reagents per reaction as mentioned in Table 5
- Transfer 14 µL of the Mastermix to each reaction tube
- Add 11 µL minus (volume for 5 µg of the nuclear extract) of 1x Transcription Buffer to each reaction tube.
- Add 5 µg of the nuclear extract to each reaction tube.
- Gently tap the reaction tube to mix the contents inside and pulse centrifuge the tubes after mixing to ensure no reaction material is stuck to the wall of the tubes.
- Incubate the reactions at 30 °C for 30 min.
- Immediately stop the reaction by adding 400 µL of RLT buffer provided by the RNA extraction kit.
NOTE: At this point, it is safe to stop. Samples can be stored at -80 °C until further clean up with the RNA extraction kit.
7. RNA clean up
- Prepare DNase I stock solution using the RNase-Free DNase set provided in the RNA extraction kit. Dissolve the lyophilized DNase I in 550 µL of RNase-free water. Gently mix the solution. Do not vortex . Aliquot the DNase I solution into 10 µL single-use tubes and store the aliquots at -20 °C.
- Prepare 70% and 80% ethanol using molecular grade ethanol and RNase-free water.
- Move the samples and reagents to an RNase-free workspace before beginning the clean-up.
- Add 400 µL of 70% ethanol to each sample and gently pipette to mix well.
- Transfer 400 µL of the sample to a labeled RNA extraction spin column with a 2 mL collection tube and centrifuge the sample at 8,500 x g for 30 s. Discard the flow-through.
- Repeat step 7.5 with the remaining sample and discard the flow through.
- Add 350 µL of RW1 buffer (RNA extraction kit) to each column. Centrifuge the columns at 8,500 x g for 30 s. Discard the flow-through.
- Add 70 µL of RDD buffer (RNA extraction kit) to a single 10 µL aliquot of DNase I stock solution and mix gently. Do not vortex.
- Add the DNase I incubation mix (80 µL) directly to the spin column membrane. Incubate the columns at room temperature for 15 min.
NOTE: Be sure to add the DNase I solution to the membrane. Avoid losing part of the solution to the column wall or O-ring. - After 15 min, add 350 µL of RW1 buffer to the columns. Centrifuge for 30 s at 8,500 x g. Discard the flow-through.
- Place the columns into new 2 mL collection tubes. Add 500 µL of RPE buffer (RNA extraction kit) to the spin column. Centrifuge the columns at 8,500 x g for 30 s to wash the membrane. Discard the flow-through.
- Add 500 µL of 80% ethanol to each column and centrifuge the columns at 8,500 x g for 30 s. Discard the flow-through.
- Place the columns into new 2 mL collection tubes. Leaving the column lids open, centrifuge the columns at 17,900 x g for 5 min. Discard the flow-through.
- Place the columns into labeled 1.7 mL tubes. Add 17 µL of RNase-free water directly to the center of the spin column membrane. Let the columns rest at room temperature for 1 min. Centrifuge the columns at 17,900 x g for 1 min.
- Discard the column and keep the 1.7 mL tube with the freshly purified RNA sample.
8. DNA digestion
- Preheat two heat blocks at 37 °C and 65 °C.
- Add 2 µL of 10x Reaction Buffer to each sample.
- Add 1 µL (1 MBU) of DNase to each sample.
- Set a pipette to 10 µL and pipette the new solution up and down to mix gently. Do not vortex.
- Incubate the RNA samples at 37 °C for 30 min to digest any remaining DNA.
- Inactivate the DNase by incubating the samples on the heat block at 65 °C for 10 min.
NOTE: It is safe to stop at this step and store the RNA at -80 °C to perform reverse transcription later.
9. Reverse transcription
- When programing the thermocycler, add an additional 1 h, 37 °C step before incubation to preheat the thermocycler. Run the program with the 'preheat step' while preparing the samples and let the thermocycler reach 37 °C. When the samples for reverse transcription are ready, skip the 37 °C preheat step and proceed to the actual incubation step (Table 6). If the thermocycler does not have a skip feature, add a 1 min 37 °C step before incubation. Allow the thermocycler to heat to the proper temperature before pausing the system and adding the prepared samples.
- Prepare a 10 µM working solution of transcription reverse primer in RNase-free H2O and store it on ice.
- Thaw on ice, 10x buffer from the reverse transcription kit, dNTP mix (5 mM each dNTP), RNase Inhibitor, and the reverse transcriptase.
- Prepare a Mastermix (per reaction) by adding the constituents mentioned in Table 7 into a clean 0.2 mL PCR tube.
- Aliquot 18 µL of the Mastermix into new 0.2 mL PCR tubes for each sample.
- Add 2 µL of the DNase treated RNA for each sample into their respectively labeled tubes.
- Incubate the samples at 37 °C for 1 h in a preheated thermocycler.
- Move the samples to the -20 °C for storage after reverse transcription or proceed directly to the next step.
NOTE: It is safe to stop at this step; store the cDNA at -20 °C for later use.
10. Specific product amplification
- Thaw on ice: cDNA, 10 µM transcription forward and 10 µM transcription reverse primers, and the PCR 2x Premix A.
NOTE: Aliquot the PCR 2x Premix A into smaller volumes to reduce the thaw time. - Create a Mastermix (per reaction) by adding the constituents mentioned in Table 8 into a clean 0.2 mL PCR tube.
- Aliquot 24 µL of Mastermix for each sample into clean labeled 0.2 mL PCR tubes.
- Add 1 µL of cDNA of each sample into their respective tubes.
- Incubate the samples in the thermocycler using the program conditions mentioned in Table 9
- After the incubation, store the PCR products at 4 °C or -20 °C for longer-term storage.
11. Gel analysis
- Use 1x TAE buffer to prepare a gel that contains 2% w/v of agarose and 1x gel stain.
- Run the PCR products at 50 V and 300 mA for 1 h or until there is a clear band separation.
- Image the gel using the automated exposure program preloaded in the gel imager.
Representative Results
Following the outlined steps should yield functional nuclear extract (Figure 1), deviation in the grinding or wash steps can lead to poor activity or low yields. If functional C. elegans nuclear extract is obtained, it will transcribe the region downstream of the CMV promoter on the DNA template when added to the previously described in vitro assay. The resulting RNA transcript can be purified from the nuclear proteins and DNA template using conventional methods. Without the template DNA, reverse transcription and subsequent PCR product can only be the result of the RNA transcribed by the nuclear extract. The PCR products can be visualized on an agarose gel, the intensity of the DNA band can be indicative of the quality of nuclear protein and RNA isolation. A weak band intensity can be caused by the inactivation of the nuclear extract either by heat or poor buffer preparation. An excessively strong band intensity can be the result of DNA contamination either due to poor RNA purification or improper DNase digestion. Consistent and successful nuclear isolations will produce bands of similar intensities, and both transcriptional and PCR negative controls should have no visible PCR product (Figure 2).
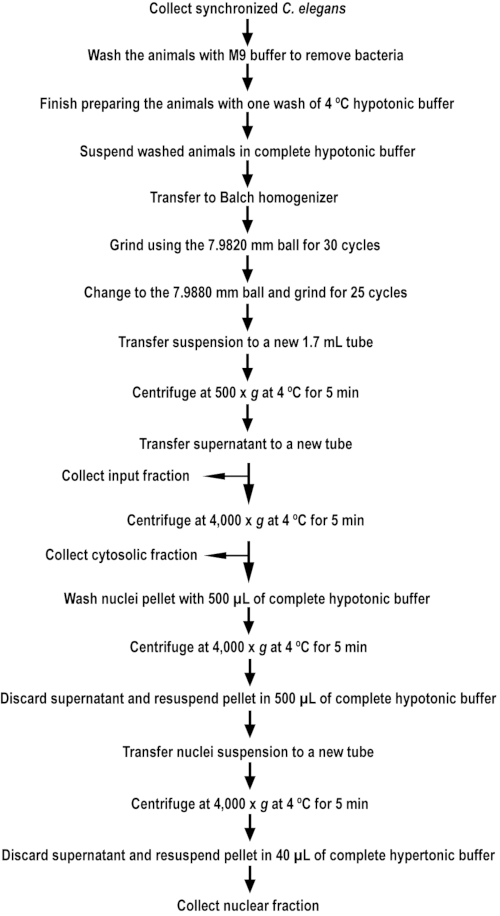
Figure 1. An outline of nuclear extract isolation. The flowchart outlines the major steps for isolating nuclear extract from C. elegans. Please click here to view a larger version of this figure.
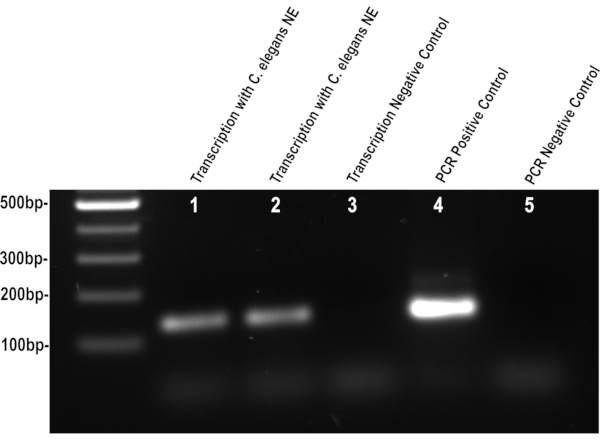
Figure 2. C. elegans nuclear extract retains its activity. The gel image shows the transcription products of C. elegans L4 larvae nuclear extract using the CMV promoter DNA template. Successful isolation of active nuclear proteins will result in a 132 bp PCR product after in vitro transcription, as seen in lanes 1 and 2. Unsuccessful isolation will result in a weak band or the absence of a PCR product similar to lane 3. This visualization of the transcription activity via PCR amplification is a simple way to assess the quality of the nuclear extraction isolation. The positive PCR control is produced by adding the CMV promoter DNA template to the PCR reaction, and the negative control is lacking the template DNA. Please click here to view a larger version of this figure.
| Nematode Growth Media Plates | |
| Agar | 20.4 g |
| Sodium Chloride | 2.8 g |
| Bacto peptone | 2.3 g |
| dH2O | 975 mL |
| Autoclave at 121 °C for 30 min | |
| Allow the media to cool to 50 °C before adding the following | |
| 1 M CaCl2 (Sterile) | 1 mL |
| 1 M MgSO4 (Sterile) | 1 mL |
| 10,000 units/mL Nystatin (Sterile) | 3 mL |
| 5 mg/mL Cholesterol in 95% Ethanol (Filter Sterilized) | 1 mL |
| 1M KPO4 pH 6.0 (Sterile) | 25 mL |
Table 1.
| M9 Buffer | |
| KH2PO4 | 3 g |
| Na2HPO4 | 6 g |
| NaCl | 5 g |
| dH2O | 1,000 mL |
| Autoclave at 121 °C for 15 min | |
| 1 M MgSO4 (Sterile) | 1 mL (add after autoclaving) |
| S-basal Buffer | |
| KH2PO4 | 6 g |
| K2HPO4 | 1 g |
| NaCl | 5.85 g |
| dH2O | 1,000 mL |
| Autoclave at 121 °C for 15 min | |
| Cholesterol 5 mg/mL (Sterile) | 1 mL (add after autoclaving) |
Table 2.
| Hypotonic Buffer | ||
| Stock Solution | Volume | Final concentration |
| 1 M HEPES KOH pH 7.6 | 7.5 mL | 15 mM |
| 1 M KCl | 5.0 mL | 10 mM |
| 1 M MgCl2 | 2.5 mL | 5 mM |
| 0.5 M EDTA | 0.1 mL | 0.1 mM |
| 1 M Sucrose | 175 mL | 350 mM |
| dH2O | 309.9 mL | |
| Filter sterilize | ||
Table 3.
| Hypertonic Buffer | ||
| Stock Solution | Volume | Final Concentration |
| 1 M HEPES KOH pH 7.6 | 7.5 mL | 15 mM |
| 1 M KCl | 200 mL | 400 mM |
| 1 M MgCl2 | 2.5 mL | 5 mM |
| 0.5 M EDTA | 0.1 mL | 0.1 mM |
| 10% Tween 20 | 5 mL | 0.10% |
| 50% Glycerol | 100 mL | 10% |
| dH2O | 184.9 mL | |
| Filter sterilize | ||
Table 4.
| MgCl2, 50 mM | 1.5 µL |
| rNTP mix, 10 mM each | 1.0 µL |
| Template DNA, 25 ng/µL | 4.0 µL |
| RNase-free H2O | 7.5 µL |
Table 5.
| Step | Temp | Time | Cycle number |
| Preheat | 37 °C | 60 min | 1x |
| Reverse Transcription | 37 °C | 60 min | 1x |
| Hold | 10 °C | 1x |
Table 6.
| 10x Reverse Transcription Buffer | 2.0 µL |
| dNTP Mix (5 mM each dNTP) | 2.0 µL |
| Transcription Reverse Primer (10 µM) | 2.0 µL |
| RNase Inhibitor | 1.0 µL |
| Sensiscript Reverse Transcriptase | 1.0 µL |
| RNase-free H2O | 10.0 µL |
Table 7.
| RNase-Free H2O | 6.25 µL |
| Transcription Forward Primer (10 µM) | 2.5 µL |
| Transcription Reverse Primer (10 µM) | 2.5 µL |
| PCR 2X Premix A | 12.5 µL |
| PCR Enzyme Mix | 0.25 µL |
Table 8.
| Step | Temp | Time | Cycle number |
| Initial Denature | 92 °C | 60 s | 1x |
| Denature | 92 °C | 30 s | |
| Anneal | 59 °C | 30 s | 35x |
| Extension | 72 °C | 30 s | |
| Hold | 10 °C | 1x |
Table 9.
Discussion
C. elegans is an appealing model organism to study the eukaryotic transcription system because of its low-cost maintenance and the ease of genetic manipulation. Here a protocol for consistent isolation of functionally active nuclear extract from L4 C. elegans is described. Although this protocol focused on visualizing transcription activity, the cDNA produced post-transcriptionally can be quantified using RT-qPCR to obtain a more precise measurement of the transcription activity8. This method of isolating nuclear proteins from C. elegans can help expand the study of the eukaryotic transcription machinery. Since C. elegans is not a culture of cells in a dish or a colony of yeast but rather a free-roaming animal, isolating and researching its nuclear extract may give a clearer insight into how the transcriptional machinery may change over time or in various environments. This allows researchers to take advantage of C. elegans low cost and resilience. C. elegans, unlike other model organisms or cell cultures, is much more forgiving when bacterial or yeast contamination appears. A population of C. elegans can be easily cleaned of contamination using established protocols, saving time and effort when contamination does occur9. Overall, using nuclear extract from C. elegans for biochemical assays can be a more affordable and flexible option as compared to purchasing nuclear extract from a vendor or dealing with less forgiving model organisms.
While this protocol is relatively simple, there are still critical steps that require special attention for the successful isolation of the nuclear extract. It is important that during the preparation of the complete hypotonic and hypertonic buffers, the two solutions are clearly labeled and separated. If the buffers are switched at any point during the isolation, this could lead to inactivation of the nuclear proteins or poor fractionation of the cytosolic proteins from the nuclear proteins. Isolated nuclear proteins should also, if necessary, be diluted in hypertonic buffer, not water or any other solution. The high salt concentration helps preserve the activity of the proteins, and a hypotonic solution can kill this activity10.
Another challenge that may arise during the grinding portion of this protocol comes from debris adhering to the surface of the tungsten balls. While it is stated that the balls should be washed and dried after every grinding cycle, the material will attach to the smooth surface of the balls. This material usually appears as rust-colored rings around the circumference of the balls and is thick enough to block the gap between the ball and the wall of the grinding chamber. This blockage is noticeable as it becomes more and more difficult to push the animals through the grinder, which can eventually lead to muscle injury or rupturing of the syringe. If the tungsten balls begin to show discoloration, soak them in hot water for 5 min then clean the surface with a new scouring pad. Avoid using acidic or basic cleaning solutions. After gently polishing, the tungsten balls should return to their original shine, and it will be noticeably easier to grind samples.
This protocol is designed to isolate whole nuclear extract from C. elegans. It has not been tested for use on other model organisms. Nuclear extract from other organisms may require different buffers, and the CMV promoter may not be enough to drive transcription in other non-mammalian samples. The nuclear extract collected using this method is also not tissue or cell-specific; any transcription activity measured using this method looks at the animals as a whole which may hide the subtle changes between tissues.
Future uses of this protocol could be measuring the DNA repair or replication machinery of C. elegans after DNA damage. The cytosolic fraction collected during the isolation process could be utilized to measure the amount of soluble proteins and quantify the activity of these proteins in a manner similar to measuring transcription.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by an NIH MIRA grant (R35GM124678 to J. S.).
Materials
| Consumables and reagents | |||
| 0.2 mL 8-Strip Tubes & Flat Strip Caps, Clear | Genesee Scientific | 24-706 | |
| 0.2 mL Individual PCR tubes | Genesee Scientific | 24-153G | |
| 1.7 mL sterile microtubes | Genesee Scientific | 24-282S | |
| 100% absolute molecular grade ethanol | Fisher Scientific | BP2818 | |
| 100% ethanol, Koptec | Decon Labs | V1001 | |
| 10 mL serological pipet | VWR international | 89130-898 | |
| 150 mm petri plates | Tritech Research | T3325 | |
| 15 mL conical centrifuge tubes | Genesee Scientific | 28-103 | |
| 20 mL plastic syringes | Fisher Scientific | 14955460 | |
| 2 mL Norm-Ject syringes | Henke-Sass Wolf GmbH | 4020 | |
| 500 mL vacuum filter cup 0.22 µm PES, Stericup Millipore Express Plus | Millipore Sigma | SCGPU10RE | |
| 50 mL conical centrifuge tubes | ThermoFisher Scientific | 339652 | |
| 50 mL serological pipet | VWR international | 89130-902 | |
| 5 mL serological pipet | VWR international | 89130-896 | |
| Agar, Criterion | VWR International | C7432 | |
| Agarose | Denville Scientific | CA3510-6 | |
| Alcohol proof marker | VWR International | 52877-310 | |
| Bacto peptone | VWR International | 90000-264 | |
| Caenorhabditis elegans | CGC | N2 | |
| Calcium dichloride | Millipore Sigma | C4901 | |
| Cholesterol | Millipore Sigma | C8667 | |
| Control DNA temple cloning primers, Forward 5’- ctc atg ttt gac agc tta tcg atc cgg gc -3’ | |||
| Control DNA temple cloning primers, Forward 5’- aca gga cgg gtg tgg tcg cca tga t -3’ | |||
| Deionized water | |||
| Dithiothreitol | Invitrogen | 15508-013 | |
| DNA gel stain, SYBR safe | Invitrogen | S33102 | |
| DNA ladder mix, O’gene ruler | Fisher Scientific | SM1173 | |
| DNA Loading Dye, 6x TriTrack | Fisher Scientific | FERR1161 | |
| DNase, Baseline-ZERO | Lucigen | DB0715K | |
| Dry ice | |||
| Escherichia coli OP50 strain | CGC | OP50 | |
| Glacial acetic acid | Fisher Scientific | A38 | |
| Glycerol | Millipore Sigma | G6279 | |
| HeLa nuclear extract in vitro transcription system, HeLaScribe | Promega | E3110 | |
| Hepes Solution, 1 M Gibco | Millipore Sigma | 15630080 | |
| Hydrochloric acid 37% | Millipore Sigma | P0662 | |
| Hypochlorite bleach | Clorox | ||
| LB Broth | Millipore Sigma | L3022 | |
| Magnesium dichloride | Millipore Sigma | M8266 | |
| Magnesium Sulfate | Millipore Sigma | M7506 | |
| Medium weigh dishes | Fisher Scientific | 02-202-101 | |
| microscope slides, Vista vision | VWR International | 16004-368 | |
| molecular grade water, Hypure | Hyclone Laboratories | SH30538 | |
| Nystatin | Millipore Sigma | N1638 | |
| PCR system, FailSafe with premix A | Lucigen | FS99100 | |
| Potassium chloride | Millipore Sigma | P39111 | |
| Potassium phosphate dibasic | Millipore Sigma | P3786 | |
| Potassium phosphate monobasic | Millipore Sigma | P0662 | |
| Protease inhibitor, Halt single use cocktail 100x | ThermoFisher Scientific | 78430 | |
| protein assay kit, Qubit | ThermoFisher Scientific | Q33211 | |
| reverse transcription kit, Sensiscript | Qiagen | 205211 | |
| RNA extraction kit RNeasy micro kit | Qiagen | 74004 | |
| RNase Inhibitor | Applied Biosystems | N8080119 | |
| Sodium Chloride | VWR International | BDH9286-12KG | |
| Sodium hydroxide | Millipore Sigma | 1-09137 | |
| Sterile syringe filter with 0.2 µm Polyethersulfone membrane | VWR international | 28145-501 | |
| Sucrose | VWR International | 200-334-9 | |
| transcription primers, Forward 5’- gcc ggg cct ctt gcg gga tat -3’ | |||
| transcription primers, Reverse 5’- cgg cca aag cgg tcg gac agt-3’ | |||
| Tris-Base | Fisher Scientific | BP152 | |
| Tween20 | Millipore Sigma | P2287 | |
| Equipment | |||
| -20 °C incubator | ThermoFisher Scientific | ||
| 20 °C incubator | ThermoFisher Scientific | ||
| 37 °C incubator | Forma Scientific | ||
| 4 °C refrigerator | ThermoFisher Scientific | ||
| -80 °C freezer | Eppendorf | ||
| Autoclave | Sanyo | ||
| Balch homogenizer, isobiotec cell homogenizer | Isobiotec | ||
| Benchtop Vortexer | Fisher Scientific | 2215365 | |
| Centrifuge, Eppendorf 5418 R | Eppendorf | 5401000013 | |
| Centrifuge, VWR Clinical 50 | VWR International | 82013-800 | |
| Dissection microscope, Leica M80 | Leica Microsystems | ||
| Fluorometer, Qubit 2.0 | Invitrogen | Q32866 | |
| Gel imaging system, iBright FL1500 | ThermoFisher Scientific | A44241 | |
| Gel system | ThermoFisher Scientific | ||
| Heat block | VWR International | 12621-048 | |
| Microcentrifuges, Eppendorf 5424 | Eppendorf | 22620401 | |
| PIPETBOY acu 2 | Integra | 155017 | |
| Pipette L-1000 XLS+, Pipet-Lite LTS | Rainin | 17014382 | |
| Pipette L-10 XLS+, Pipet-Lite LTS | Rainin | 17014388 | |
| Pipette L-200 XLS+, Pipet-Lite LTS | Rainin | 17014391 | |
| Pipette L-20 XLS+, Pipet-Lite LTS | Rainin | 17014392 | |
| Rocking platform | VWR International | ||
| Thermocycler, Eppendorf Mastercycler Pro | Eppendorf | 950030010 |
References
- Corsi, A. K., Wightman, B., Chalfie, M. A Transparent window into biology: A on Caenorhabditis elegans. 遗传学. 200 (2), 387-407 (2015).
- Blackwell, T. K., Walker, A. K. Transcription mechanisms. WormBook: The Online Review of C. elegans Biology. , 1-16 (2006).
- Page, A. P., Johnstone, I. L. The cuticle. WormBook: The Online Review of C. elegans Biology. , 1-15 (2007).
- Lichtsteiner, S., Tjian, R. Cloning and properties of the Caenorhabditis elegans TATA-box-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 90 (20), 9673-9677 (1993).
- Lichtsteiner, S., Tjian, R. Synergistic activation of transcription by UNC-86 and MEC-3 in Caenorhabditis elegans embryo extracts. EMBO Journal. 14 (16), 3937-3945 (1995).
- Shan, G., et al. Isotope-labeled immunoassays without radiation waste. Proceedings of the National Academy of Sciences of the United States of America. 97 (6), 2445-2449 (2000).
- Voss, C., et al. A novel, non-radioactive eukaryotic in vitro transcription assay for sensitive quantification of RNA polymerase II activity. BMC Molecular Biology. 15, 7 (2014).
- Wibisono, P., Liu, Y., Sun, J. A novel in vitro Caenorhabditis elegans transcription system. BMC Molecular and Cell Biology. 21 (1), 87 (2020).
- Stiernagle, T. Maintenance of C. elegans. WormBook: The Online Review of C. elegans Biology. , 1-11 (2006).
- Zbacnik, T. J., et al. Role of buffers in protein formulations. Journal of Pharmaceutical Sciences. 106 (3), 713-733 (2017).
