We have successfully used this protocol to generate mRNA encoding for FLAG-tagged NEMO and IKKβ variants for transfection of primary macrophages16. The plasmids encoding for FLAG-tagged wild-type (NEMOWT) and C54/347A mutant NEMO (NEMOC54/374A) (see the Table of Materials) already contain a T7 promotor in the correct orientation (Figure 1A). Thus, we only had to linearize the plasmids to generate DNA templates for in vitro transcription. To this end, 10 µg of plasmid DNA were digested with 5 µL Xbal resulting in linearization of the plasmid due to a single cut 3' of the stop codon. Complete linearization of the plasmid DNA was verified by agarose gel electrophoresis (Figure 1B).
The plasmids encoding for FLAG-tagged wild-type (IKKβWT) and S177/181E mutant IKKβ (IKKβS177/181E) do not contain a T7 promotor. Therefore, we have attached a T7 promotor by PCR to generate the DNA templates for in vitro transcription (Figure 2A). Generation of a specific PCR product, i.e., a single product of correct size, was verified by agarose gel electrophoresis (Figure 2B).
After in vitro transcription using the respective DNA templates, we verified generation of a single mRNA product of correct size and poly(A) tailing by agarose gel electrophoresis under denaturing conditions (Figure 3).
To verify that mRNA generated using this protocol does enable the transfection of primary macrophages (see Figure 4A,B for flow cytometric analyses of immunomagnetic enrichment of PM and differentiation status of BMDM), we have generated mRNA encoding for eGFP (Figure 5A-C) and analyzed transfection efficiency by flow cytometry (Figure 6A). 100,000 PM or 50,000 BMDM per well of an untreated microtiter plate were transfected with 50, 100 or 200 ng of eGFP mRNA for 6, 9 or 24 h. In both PM and BMDM, high levels of eGFP expression could be detected at 6 h after transfection. Transfection rate increased with the amount of transfected mRNA and was highest for 200 ng mRNA (Figure 6B,C). For PM, transfection rate reached about 50-65% at 6 to 9 h after transfection (Figure 6B). At 24 h after transfection, transfection rate was substantially lower indicating expiring eGFP expression. Thus, PM should not be transfected overnight. For BMDM, transfection rate reached about 80-85% (Figure 6C). The drop in transfection rate after 24 h was much less pronounced in BMDM. Thus, BMDM can be transfected overnight. In both PM (Figure 6D,E) and BMDM (Figure 6F,G), expression level of eGFP in transfected cells increased in a time- and dose-dependent manner. Importantly, the transfection procedure did not induce lytic or apoptotic cell death as there was no increase in propidium iodide-positive (PI) or annexin V-positive macrophages after transfection (Figure 7A,B).
Transfection efficiency of the mRNAs encoding for FLAG-tagged NEMO or IKKβ variants was analyzed by immunofluorescence microscopy. 300,000 PM per well of a 12-well plate were transfected with 300 ng of respective mRNA for 6 h. Transfection rate was about 60% for NEMO mRNAs and about 55% for IKKβ mRNAs (Figure 8A,B). Thus, their transfection rates were similar to that of eGFP mRNA indicating a general transfection rate of about 55% for PM.
We have also used this protocol to generate mRNA encoding for Cre recombinase. Transfection of 400,000 BMDM from NEMOflox/flox mice17 per well of a 12-well plate with 400 ng of Cre recombinase mRNA resulted in almost complete depletion for NEMO protein after 48 h (Figure 9) indicating highly efficient transfection of the BMDM.
PM and BMDM did not secrete any detectable amounts of IL-1β, IL-6 and TNF after transfection (Figure 10A,B). Moreover, the NF-kB and MAPK signaling pathways were not activated after transfection16 indicating that the transfection procedure does not activate proinflammatory signaling. We also have not observed any functional alterations of transfected macrophages in comparison to untransfected macrophages16.
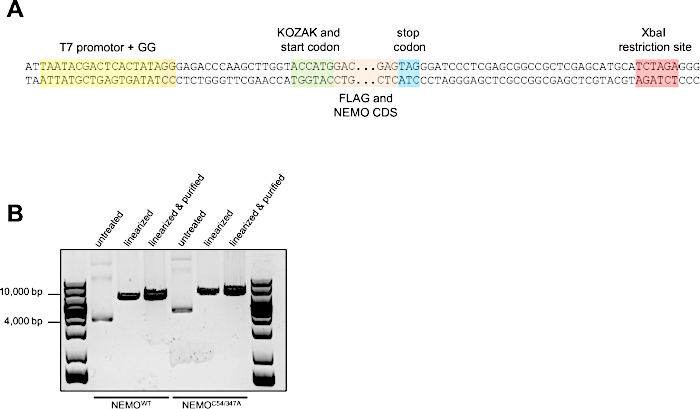
Figure 1: Linearization of NEMO-encoding plasmids already containing a T7 promotor in the correct orientation. (A) Sequence excerpt of the plasmid encoding for FLAG-tagged NEMOWT or NEMOC54/347A. A T7 promotor in correct orientation and close to the KOZAK sequence and start codon is already present. The T7 promotor region, the coding sequence (CDS) for the FLAG tag and NEMO, the start and stop codons and the restriction site for linearization with XbaI are color-coded. (B) Representative 1% agarose gel showing the plasmids before and after linearization with Xbal; 1 µL of untreated, linearized or purified linearized plasmid DNA was loaded per lane. Please click here to view a larger version of this figure.
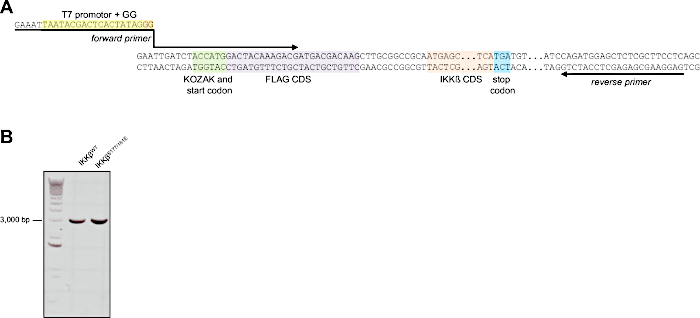
Figure 2: T7 promotor attachment to IKKß-encoding plasmids by PCR. (A) Sequence excerpt of the plasmid encoding for FLAG-tagged IKKβWT or IKKβS177/181E. The plasmids do not contain a suitable T7 promotor, which is therefore attached by PCR. Location, orientation and sequence of the forward primer (containing the T7 promotor sequence to be added) and the reverse primer are indicated by the arrows. The T7 promotor region, the CDS for the FLAG tag and IKKβ and the start and stop codons are color-coded. (B) Representative 1% agarose gel verifying generation of a single PCR product of correct size using the primers indicated above. 1 µL of the purified PCR product was loaded per lane. Please click here to view a larger version of this figure.
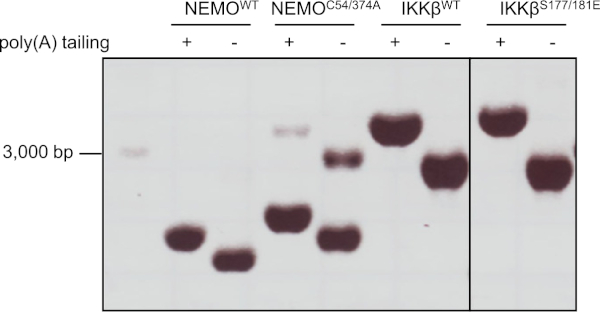
Figure 3: mRNA synthesis from NEMO- and IKKβ-encoding DNA templates. (A) Representative denaturing 1.2% agarose gel containing 0.7% formaldehyde showing the purified mRNA of NEMO and IKKβ constructs before and after poly(A) tailing. 2 µL of NEMOWT, NEMOC54/347A, IKKβWT and IKKβS177/181E mRNA were loaded per lane. A 32 µL ssRNA ladder was loaded for RNA length determination. Please click here to view a larger version of this figure.
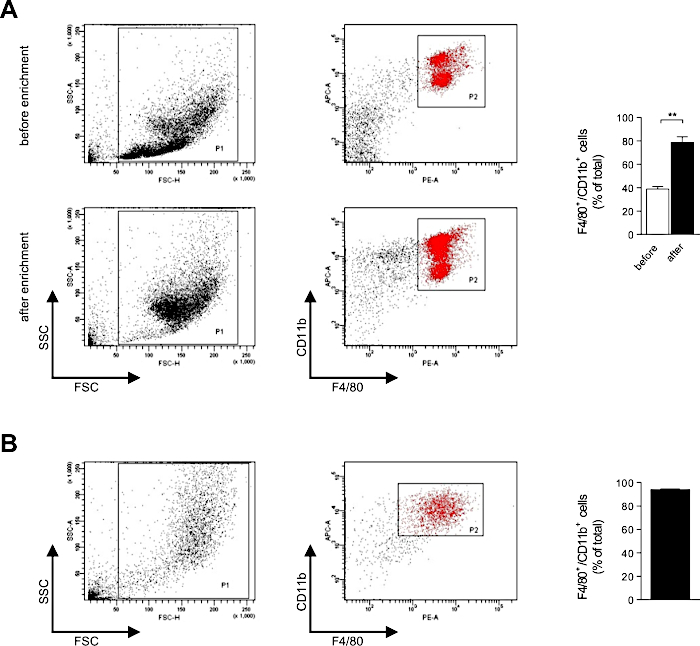
Figure 4: Flow cytometric analyses of the immunomagnetic enrichment of PM and the differentiation status of BMDM. (A) The percentage of F4/80+/CD11b+ PM in the peritoneal lavage before and after immunomagnetic enrichment was analyzed by flow cytometry. (B) Expression of F4/80 and CD11b by BMDM after 6 days of differentiation was verified by flow cytometry. 10,000 or 5,000 cells were counted per sample, respectively. Data are shown as mean ± SEM of n = 3 independent experiments, each. * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001 by Student's t-test. Please click here to view a larger version of this figure.
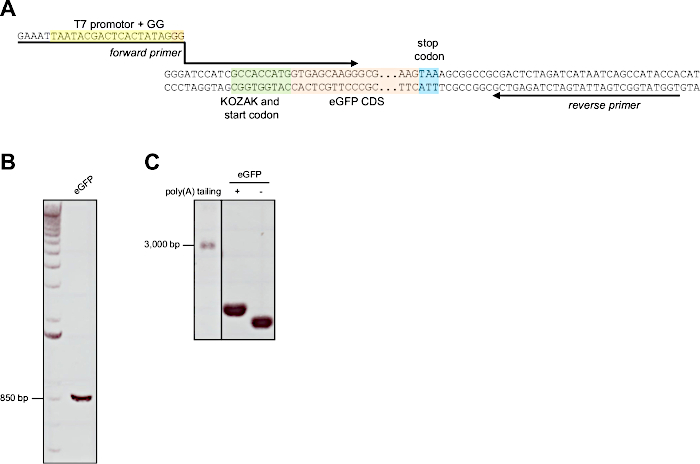
Figure 5: mRNA synthesis with poly(A) tailing of eGFP. (A) Sequence excerpt of the plasmid encoding for eGFP. The plasmid does not contain a suitable T7 promotor, which is therefore attached by PCR. Location, orientation and sequence of the forward primer (containing the T7 promotor sequence to be added) and the reverse primer are indicated by the arrows. The T7 promotor region, the CDS for eGFP and the start and stop codons are color-coded. (B) Representative 1% agarose gel showing the purified amplicon after PCR using the primers indicated above. 1 µL of the purified PCR product was loaded per lane. (C) Representative denaturing 1.2% agarose gel containing 0.7% formaldehyde showing the purified mRNA of eGFP before and after poly(A) tailing. 2 µL of eGFP mRNA were loaded per lane. A 32 µL ssRNA ladder was loaded for RNA length determination. Please click here to view a larger version of this figure.
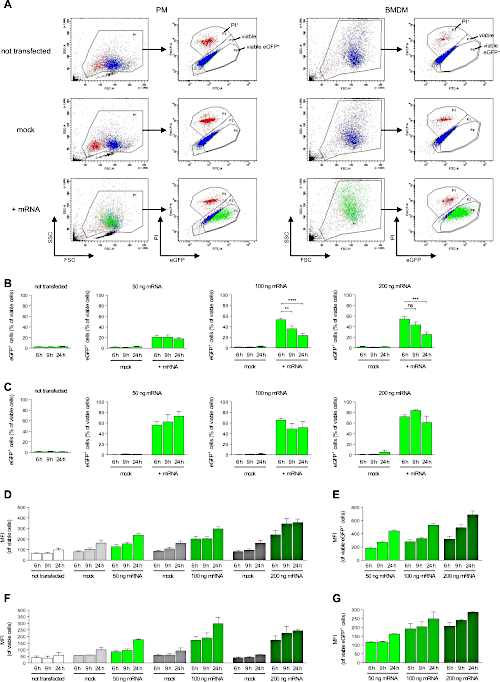
Figure 6: Highly efficient transfection of both peritoneal macrophages and BMDM with eGFP mRNA. Macrophages were incubated for 6, 9 or 24 h with 50, 100 or 200 ng of eGFP mRNA complexed to jetMESSENGER or with jetMESSENGER alone (mock) and then analyzed by flow cytometry. (A) Gating strategy used to define the populations of viable macrophages, viable eGFP+ macrophages and PI+ (i.e., dead) macrophages. Representative data for transfection with 200 ng mRNA for 6 h are shown. (B,C) Transfection rates of (B) PM and (C) BMDM were determined by analyzing the percentage of viable eGFP-positive cells by flow cytometry (n = 5-7 and n = 4-5 independent experiments, respectively). (D-G) Expression levels of eGFP were determined by analyzing the mean fluorescence intensity (MFI) of viable (D) PM and (F) BMDM and of the viable and eGFP-positive subpopulation of (E) PM and (G) BMDM (n = 5-7 and n = 4-5 independent experiments, respectively). 10,000 cells were counted per sample. Data are shown as mean ± SEM. n.s. = not significant; * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001 by Student's t-test. Please click here to view a larger version of this figure.
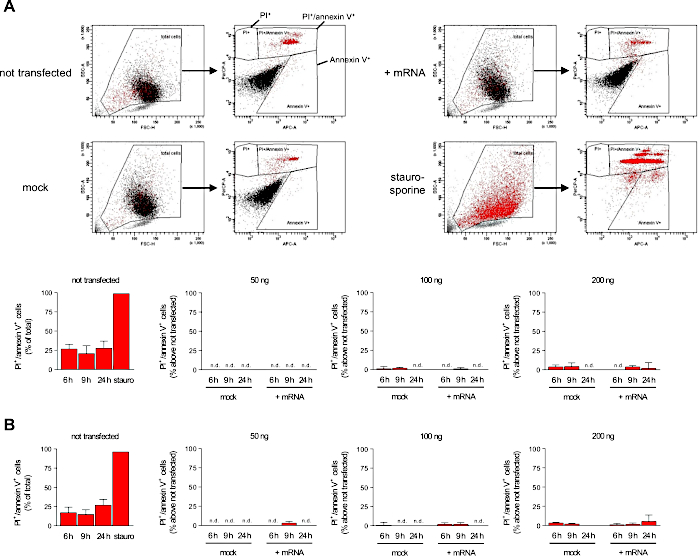
Figure 7: Transfection does not induce lytic or apoptotic cell death. Transfection-induced cell death of (A) PM and (B) BMDM was determined by analyzing the percentage of PI-positive and annexin V-positive cells (n = 5-7 and n = 4-5 independent experiments, respectively). The percentage of dead macrophages present in untransfected samples (some degree of cell death was caused by physical detachment of macrophages from the wells despite usage of non-treated plates) was subtracted from that in the respective transfected samples to only take into account cell death induced by the transfection procedure. The gating strategy used to define the populations of PI+, annexin V+ and PI+/annexin V+ macrophages is shown for a representative data set of PM transfected with 200 ng mRNA for 6 h are shown. Staurosporine (50 µM for 1 h) was used as positive control. 10,000 cells were counted per sample. Data are shown as mean ± SEM. n.d. = not detectable. Please click here to view a larger version of this figure.
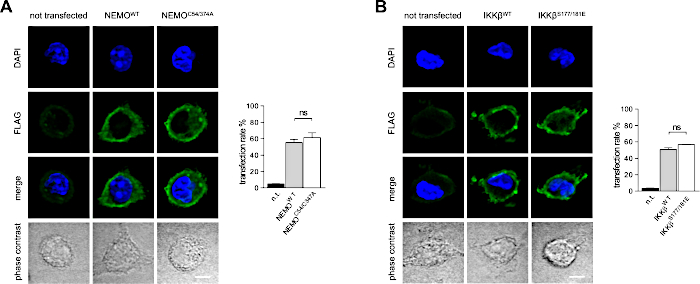
Figure 8: Transfection rates using mRNA encoding for NEMO and IKK constructs. Transfection rates using mRNA encoding for (A) NEMO constructs and (B) IKK constructs were quantified by immunofluorescence microscopy (n = 4 independent experiments). Scale bar = 4 µm. Data are shown as mean ± SEM. n.t. = not transfected, n.s. = not significant; * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001 by Student's t-test. Please click here to view a larger version of this figure.
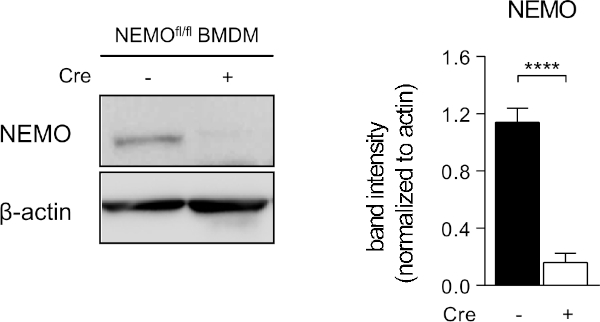
Figure 9: Transfection of NEMOfl/fl BMDM with mRNA encoding for Cre recombinase results in almost complete deficiency for NEMO. BMDM from NEMOfl/fl mice were transfected with mRNA encoding Cre recombinase for 48 h. Deficiency for NEMO protein as a result of Cre-mediated knockout was assessed by western blot using specific antibodies recognizing NEMO or β-actin and quantified by densitometry (n = 5 independent experiments). Data are shown as mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001 by Student's t-test. Please click here to view a larger version of this figure.
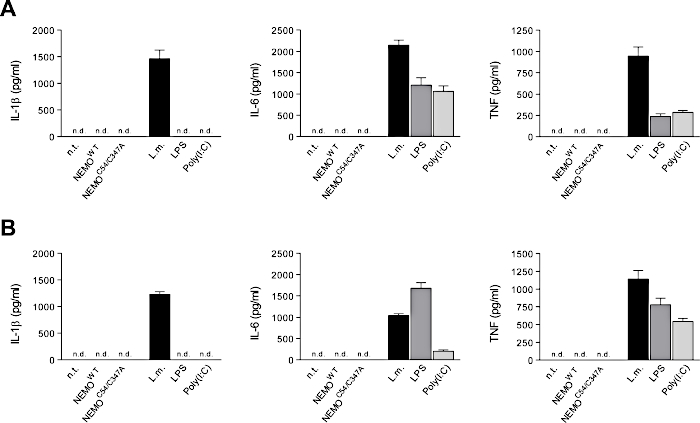
Figure 10: Transfection does not induce a proinflammatory response. (A) PM and (B) BMDM were transfected with mRNA encoding for NEMOWT or NEMOC54/347A. As positive control, macrophages were infected with Listeria monocytogenes at multiplicity of infection of 1 or stimulated with 5 µg/mL LPS or poly(I:C) for 5 h (PM) or 24 h (BMDM). Secretion of IL-1 β, IL-6, and TNF into the supernatant was quantified by ELISA (n = 3 independent experiments). Data are shown as mean ± SEM. n.t. = not transfected, n.d. = not detectable. Please click here to view a larger version of this figure.
