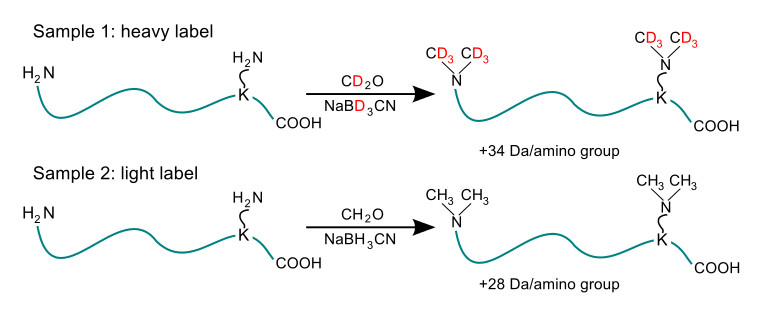
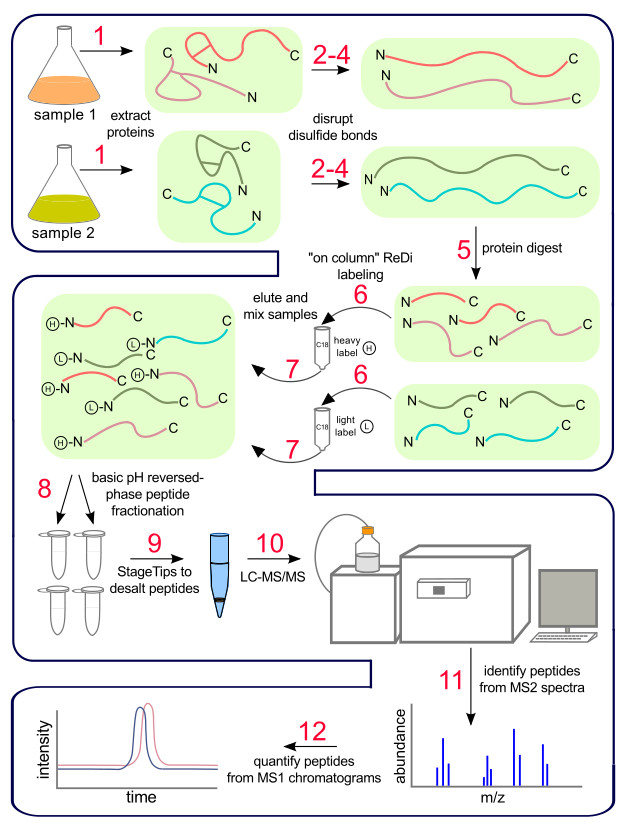
NOTE: This method was previously described12.
1. Protein Isolation
Prepare 1 mg of cellular protein by lysing cells, preferably by physical methods such as French press, bead beating, or sonication. Avoid lysozyme-mediated cell lysis because the enzyme will confound mass spectrometry measurements.
2. TCA Precipitation of Proteins
Add 1 volume trichloroacetic acid (TCA) to 4 volumes protein and chill on ice for 10 min to precipitate proteins. Centrifuge at 12,000 x g for 5 min at 4 °C and remove the supernatant. Resuspend the pellet in 1 ml of ice-cold acetone and centrifuge at 12,000 x g for 5 min at 4 °C. Remove the supernatant and invert the tube on the bench to dry the pellet for 15 min. Store protein pellets at -80 °C.
3. Denature Proteins and Reduce Disulfide Bonds
Resuspend proteins to ~2 mg/ml in 500 μl denaturation and reduction buffer (either 4 M urea or 3% SDS in 50 mM HEPES pH 8.5, 5 mM DTT). Optionally, include a protease inhibitor in the buffer. Incubate proteins for 30 min at 56 °C, followed by 10 min at room temperature.
4. Aklylate Free Sulfhydryl Groups to Irreversibly Disrupt Disulfide Bond Formation
Prepare fresh 0.3 M iodoacetamide in water. CAUTION! Iodoacetamide is highly toxic. Add 25 μl 0.3 M iodoacetamide (15 mM final concentration) to 500 μl protein and incubate for 20 min in the dark at room temperature. Quench iodoacetamide by adding 10 μl of 300 mM DTT (5 mM final DTT concentration). Store alkylated proteins at -80 °C.
5. Protein Digestion
TCA precipitate proteins (as described in step 2) and resuspend in 1 ml of 50 mM HEPES (pH 8.2), 1 M urea. Prepare a stock solution of Lysyl endoproteinase (Lys-C) in water at a concentration of 2 µg/µl and add 5 µl to the protein solution. Incubate the mixture for 16 hr at room temperature. Ensure the final Lys-C concentration is 10 ng/µl and the protein-to-LysC ratio (w/w) is 1/50 to 1/200. Resuspend 20 μg sequencing grade trypsin in 40 μl of 50 mM acetic acid, add 5 µl (10 µg trypsin) to the Lys-C digest, and incubate for 6 hr at 37 °C. Use the same protease concentration and protease-to-protein ratios for Lys-C as used for trypsin.
6. Reversed-phase Peptide Extraction
- Acidify peptides by adding trifluoroacetic acid (TFA) to a final concentration of 0.5% (pH≈2). Attach a C18 column to an extraction manifold. Use the highest possible flow rate for all steps except loading and elution of the peptides.
- Wet column with 6 ml acetonitrile (ACN). Wash column with 6 ml 80% ACN, 0.1% TFA, then equilibrate with 6 ml 0.1% TFA. Do not allow the column to run dry between steps.
- Stop vacuum pressure and load 500 μg of peptides onto the column at a flow rate of approximately 1 ml/min. Once peptides have bound to the column, restart vacuum and wash with 6 ml 0.1% TFA, then with 3 ml of citric acid buffer (0.09 M citric acid, 0.23 M Na2HPO4, pH 5.5).
Note: Higher peptide amounts can be labeled but the C18 column binding capacity should be at least two-fold higher than the peptide quantity to avoid sample loss.
7. On-column Peptide Labeling by Reductive Dimethylation (ReDi Labeling)
Perform this step under a chemical hood as hydrogen cyanide is released in low concentration during the labeling process.
- Prepare 12 ml of “light” and “heavy” ReDi buffers to methylate peptide free amines. Light ReDi buffer consists of 0.8% formaldehyde and 0.12 M sodium cyanoborohydride carrying hydrogens in their natural isotopic distributions in citric acid buffer. Heavy ReDi buffer consists of 0.8% deuterated formaldehyde and 0.12 M deuterated sodium cyanoborohydride in citric acid buffer.
- Incubate column containing peptides by adding 10 ml either light or heavy ReDi buffer to the peptide-containing columns at flow rate of 1 ml/min and repeat to ensure complete labeling. Wash column with 6 ml 0.1% TFA and then with 1 ml 0.5% acetic acid.
- Stop vacuum and elute labeled peptides first with 1 ml 40% ACN, 0.5% acetic acid, then with 1 ml 80% ACN, 0.5% acetic acid using a flow rate of approximately 0.5 ml/min. If desired, measure the labeling efficiency of individual samples by mass spectrometry before mixing heavy and light samples (see “Representative Results”). Mix 1:1 heavy and light-labeled peptide samples to be quantified by mass spectrometry.
8. Separate Peptide Mixture by Basic pH Reversed Phase (BPRP) Chromatography
Basic pH reversed phase (BPRP) chromatography to separate the peptide mixture into multiple fractions, which are independently analyzed by LC-MS/MS to increase proteome coverage.
- Fractionate the peptide mixture on a C18-HPLC column by applying a gradient of increasing ACN concentration in 10 mM ammonium bicarbonate (pH 8). Start with 5% (v/v) ACN for 5 min, increase to 35% ACN in 60 min, and then to 90% ACN in 1 min. Retain the 90% ACN for 4 min before reducing the ACN to 5% to re-equilibrate the column for 9 min. Collect 96 fractions of equal volume in a 96-well plate (A1 to H12). Monitor the fractionation using a UV detector at 220 nm while peptides are eluting off the column (10-70 min for the conditions described here).
- Combine fractions from wells A1, C1, E1, and G1 (fraction A1), from wells B1, D1, F1, and H1 (fraction B1), from wells A2, C2, E2, and G2 (fraction A2) and accordingly for the remaining fractions. Remove the solvent using a vacuum centrifuge. Resuspend peptides from fractions A1, B2, A3, B4, A5, B6, A7, B8, A9, B10, A11, and B12 in 130 µl of 1 M urea/0.5% TFA and purify using StageTips as described in step 9. Store fractions B1, A2, B3, A4, B5, A6, B7, A8, B9, A10, B11, and A12 at -20 ºC.
9. Purify Peptides by STop and Go Extraction (StageTips)
Prepare C18-StageTip7 microcolumns by packing 200 μl pipette tips with two C18 disks with an internal diameter (ID) of 1.07 mm. Put Stage Tips into Eppendorf tubes. Use a microcentrifuge to wash tips with 130 μl of methanol, then 130 ul 80% ACN, 0.5% acetic acid. Equilibrate StageTips with 130 μl 0.1% TFA. Transfer peptide mixture to StageTips and wash with 130 μl 0.1% TFA, then 40 μl 0.1% TFA, then 40 μl 0.5% acetic acid. Elute peptides first with 20 μl 40% ACN, 0.5% acetic acid, then 20 μl 80% ACN, 0.5% acetic acid. Combine eluates and dry by vacuum filtration.
10. Microcapillary LC-MS/MS
- Dissolve peptides in 1-5 μl 5% formic acid, 5% ACN to a concentration of approximately 1 μg/μl. Resolve ~1 μg peptides on a 100 μm × 20 cm C18-reversed phase HPLC column with a gradient of 6-22% ACN in 0.125% formic acid applied over 75 or 100 min at a flow rate of ~300 nl/min.
- Identify peptides by using an LTQ Orbitrap Velos12 or similar liquid chromatography-mass spectrometry platform with a mass spectrometer providing high-resolution and high mass accuracy. Operate the mass spectrometer in data-dependent mode with a full MS scan (resolution of 60,000) acquired in the Orbitrap analyzer. Generate linear ion trap MS/MS spectra for the 20 most abundant ions detected in the full MS spectrum. Set automatic gain control (AGC) targets to 1 x 106 for the full MS and 2,000 for MS/MS. Set maximum ion accumulation times to 1,000 msec for MS and 150 msec for MS/MS. Exclude fragmented peptide precursor ions from further selection from MS/MS for 20–60 sec.
11. MS/MS Data Acquisition
Identify peptides by comparing MS/MS spectra RAW files to a theoretical database with an algorithm such as SEQUEST8 using these parameters (Table 1).
Table 1. Peptide Database Search Parameters.
| General Parameters |
fully tryptic digestion with up to 2 missed cleavages |
| 25 ppm precursor ion tolerance |
| 1.0 Da fragment ion tolerance |
| Static Modifications |
+57.02146 Da on cysteine, carboxyamidomethylation |
| +28.03130 Da on lysine and the peptide N-terminus, light dimethylation label |
| Dynamic Modifications |
+15.99491 Da on methionine, oxidation |
| +6.03766 Da on lysine and the peptide N-terminus, heavy dimethylation label |
- Filter peptides to a 1% false discovery rate with a method such as the target-decoy9 strategy using a database of open reading frames in the actual and reversed orientations.
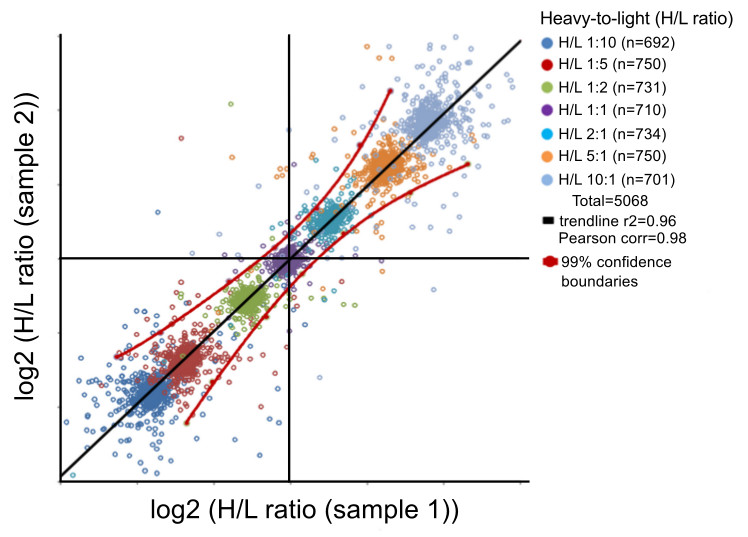
12. Peptide Quantification
Calculate the areas of heavy and light pairs of MS1 extracted ion chromatograms (MS1 peak areas) and peptide signal-to-noise (S/N) ratios10. Include peptide pairs only when their average signal-to-noise ratio is above five. Quantify relative abundance of a peptide in the two samples as the ratio of MS1 peak areas of heavy and light versions of the same peptide (MS1 peak area ratio). Calculate relative protein abundances as the median MS1 peak area ratio for all peptides in the protein.