We followed the procedure described above to record the autophosphorylation of AURKA on Thr288 using two biosensors with different spectral properties. We compared the initial GFP-AURKa-mCherry probe14 with two biosensors with different spectral properties. These two probes rely on the fluorescent donor mTurquoise2 and on a non-fluorescent acceptor (ShadowG) in one case, or a yellow acceptor (superYFP) in a second case. We then inserted the full-length sequence of AURKA within each donor/acceptor pair. To have a negative control for AURKA activation, two strategies can be pursued. First, the use a small ATP-analog (MLN8237) interferes with the binding of ATP in the kinetic pocket of the kinase and prevents its activation38. Second, the mutation of Lys162 into Met (K162M), creates a kinase-dead version of each biosensor incapable of activating14,15,39. This mutation induces the disruption of a salt bridge normally established between Lys162 and Glu181, which results in a stable opening of the kinetic pocket of the kinase and triggers its overall inactivation40. As a negative control for FRET, we used an acceptor-devoid construct (GFP-AURKA or AURKA-mTurquoise2).
After synchronizing cells in G2/M and releasing them into mitosis, we measured the lifetime of all the transfected constructs at the mitotic spindle (Figure 4). Of note, this structure was considered as a whole, and no ROIs within the spindle were analyzed. We then calculated ΔLifetime for all conditions. As expected, the lifetime of GFP-AURKA or AURKA-mTurquoise2 (the “donor-only” conditions) was close to 0, indicating that the values measured for these constructs fluctuated around the mean value (Figure 4A,4B). Conversely, the ΔLifetime values for GFP-AURKA-mCherry were statistically different from the donor-only condition, with ΔLifetime increasing of ~130 ps (Figure 4A). Similar observations were made for shadowG-AURKA-mTurquoise2 and for superYFP-AURKA-mTurquoise2, with ΔLifetime increasing of ~150 and ~220 ps from the donor-only condition, respectively (Figure 4B,4C). These data can be easily visualized in single cells with a pseudocolor Lookup Table (LUT). In this case, values of ΔLifetime around 0 are pseudocolored yellow, while more significant differences are pseudocolored red/purple. Indeed, the pixel-by-pixel LUT was closer to yellow in cells expressing the donor-only constructs, while it was more in the red/purple spectrum in cells expressing either biosensor (Figure 4A,4B). This was also observed when the GFP-AURKA-mCherry biosensor was treated with the pharmacological inhibitor MLN8237.
We then analyzed the ΔLifetime of kinase-dead biosensors. These constructs showed intermediate ΔLifetime values: ΔLifetime was significantly higher when compared to the donor-only condition (Figure 4B,4C), but it was also significantly lower than their normal counterparts (Figure 4B,4D). The comparisons with cells treated with MLN8237 or expressing kinase-dead biosensors are necessary to estimate whether ΔLifetime variations for each donor/acceptor pair are solely linked to the activation of AURKA. In the case of GFP-AURKA-mCherry, ΔLifetime variations are abolished when an AURKA-specific inhibitor is used. Conversely, ΔLifetime variations are mostly, but not exclusively linked to AURKA activation in the case of shadowG-AURKA-mTurquoise2 and of superYFP-AURKA-mTurquoise2.
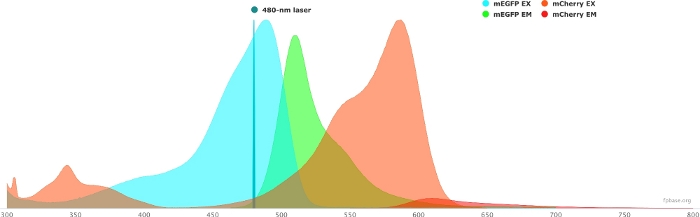
Figure 1: GFP (donor) and mCherry (acceptor) excitation and emission spectra.
Spectra were obtained and adapted from the FP base website (https://www.fpbase.org/), and adjusted to a 480 nm-laser excitation. Please click here to view a larger version of this figure.

Figure 2: Image of the experimental workspace.
(1) The control solution; (2) white laser source; (3) CCD camera; (4) microscope setup; (5) external light source/lamp for ocular screening of the sample. Please click here to view a larger version of this figure.
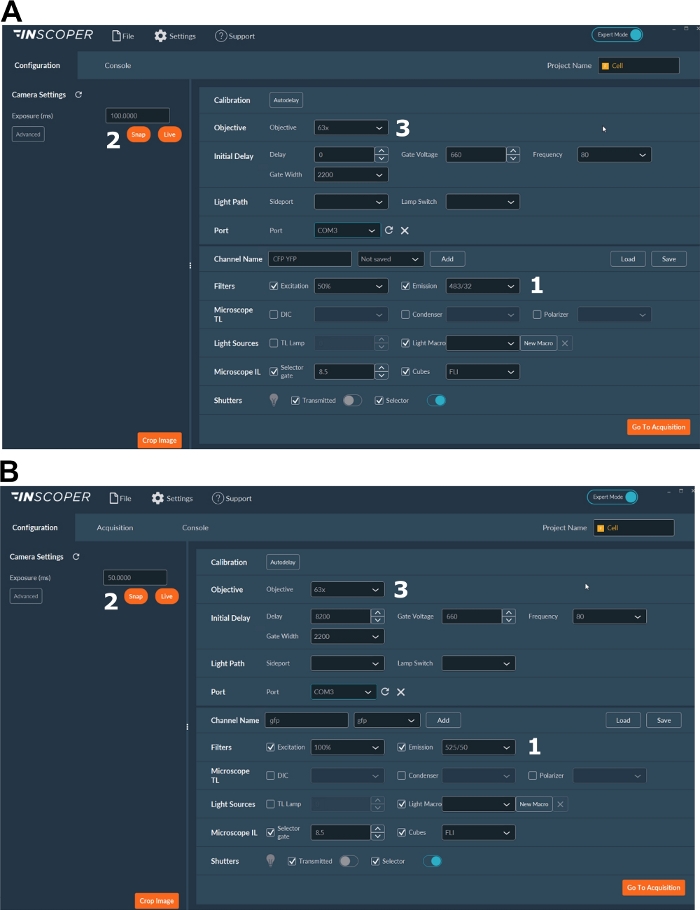
Figure 3: Representative images of the software for FLIM acquisition.
(A, B) (1) excitation and emission parameters for the donor (CFP in A, or GFP in B); (2) exposure time; (3) selection of the objective. Please click here to view a larger version of this figure.
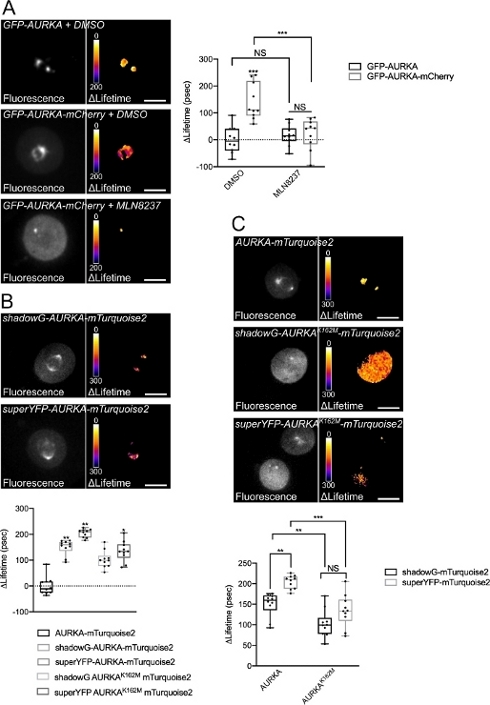
Figure 4: Representative images of AURKA FRET biosensors and their negative controls.
(A) (Micrographs) Fluorescence (green channel) and corresponding pixel-by-pixel ΔLifetime (donor only – biosensor) of U2OS cells expressing GFP-AURKA or GFP-AURKA-mCherry, synchronized at G2/M, released until the bipolar spindle is visible and then treated with DMSO or with MLN8237. ΔLifetime is illustrated with a pseudocolor scale (“Fire” lookup table). (Graph) Corresponding quantification and two-way ANOVA analysis for the indicated conditions. (B) (Micrographs) Fluorescence (cyan channel) and corresponding pixel-by-pixel ΔLifetime (donor only – biosensor) of U2OS cells expressing shadowG-AURKA-mTurquoise2 (upper panel) or superYFP-AURKA-mTurquoise2 (lower panel), synchronized at G2/M and released until the bipolar spindle is visible. ΔLifetime is illustrated with a pseudocolor scale (“Fire” lookup table). (Graph) Corresponding quantification and one-way ANOVA analysis of conditions represented in the above micrographs. (C) (Micrographs) Images of AURKA-mTurquoise2 (upper panel), shadowG-AURKA K162M-mTurquoise2 (middle panel) and superYFP-AURKA K162M-mTurquoise2 acquired and represented as in the micrographs. (Graph) Two-way ANOVA analysis for the indicated transfection conditions. The bar in boxplots represents the median; whiskers extend from the min to the max. n = 10 cells per condition of one representative experiment (of three). Individual values are represented as dots. Scale bar: 10 µm. *P < 0.05, **P < 0.01, ***P < 0.001 against each indicated condition in (A) the “AURKA-mTurquoise2” condition in (B), and against each indicated condition in (C). NS: not significant. Please click here to view a larger version of this figure.
