Automatically Generated
Unveiling Histone Proteoforms using 2D-TAU Gel Electrophoresis
Summary
This method describes a simple and fast strategy for histone proteoform characterization and investigation.
Abstract
Histones undergo various post-translational modifications (PTMs) such as methylation, acetylation, phosphorylation, acylation, and ubiquitination, which control nucleosome dynamics and determine cell fate. The nucleosome, which is the functional unit of chromatin, comprises DNA, four pairs of histones (H3, H4, H2A, and H2B) making up the globular core, and the linker histone H1, which stabilizes the chromatin structure. The amino (N)-terminal tails of the histones protrude from the globular core domains and undergo distinct PTMs that influence the chromatin landscape. Some evidence suggests that histone PTM homeostasis is crucial for preserving all physiological activities. The deregulation of histone PTMs is the primary cause of abnormal cellular proliferation, invasion, and metastasis. Therefore, developing methods for characterizing histone PTMs is crucial. Here, we describe an effective technique for isolating and analyzing histone isoforms. The method, based on the combination of two orthogonal separations, allows the enrichment of histone isoforms and the following mass spectrometry identification. The technique, originally described by Shechter et al., combines acid-urea polyacrylamide gels (TAU-GEL), which can separate basic histone proteins based on size and charge, and Sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE), which can separate proteins by molecular weight. The result is a two-dimensional map of histone isoforms, suitable for in-gel digestion followed by mass spectrometry identification and western blot analysis. The result is a two-dimensional map of histone isoforms, suitable for both in-gel digestion followed by mass spectrometry identification and western blot analysis. This proteomic approach is a robust method that allows the enrichment of a single histone isoform and the characterization of new histone PTMs.
Introduction
Cancer onset, progression, and chemoresistance are related to genetic and epigenetic events, including histone PTMs. Histones undergo a variety of PTMs that regulate chromatin dynamics and dictate cell fate1.
The nucleosome is the functional unit of chromatin and is a molecular complex of DNA and proteins that allows DNA to be packaged into the nucleus. It consists of four pairs of histones (H3, H4, H2A, and H2B) that constitute the nucleosome core, as well as the linker histone H1, which helps stabilize the chromatin structure. The histones amino (N)-terminal tails protrude from their globular core domains and undergo specific post-translational modifications that affect the chromatin landscape2.
So far, at least 23 classes of histone PTMs are known. These PTMs are covalent modifications that mainly consist of methylation, acetylation, phosphorylation, glycosylation, ubiquitylation, SUMOylation, and ADP-ribosylation3. In addition, a tight correlation between epigenome homeostasis and metabolic rewiring has recently been proposed, which explains the exponential growth of the possible number of histones PTMs4,5,6,7,8,9,10,11,12,13. Histone PTMs, deposited, erased, and transduced by modifier enzymes, are named respectively as writers, erasers, and readers. They affect the physical and chemical properties of chromatin structure, impacting gene expression profile, including the expression of tumor suppressor genes and oncogenes14,15,16.
The number of histones proteoforms increases when incorporated into the nucleosome as histone variants. These variants are non-allelic isoforms of canonical histones, and they add complexity by altering the chemical and physical properties of chromatin. In comparison to canonical histones, histone variants have functional domains that undergo unique post-translational modifications or interact with specific chromatin components, impacting the dynamics and stability of the nucleosome17.
In cancer cells, the deregulation of histone PTMs and variants affects gene expression profile, leading to abnormal cellular proliferation, invasion, metastasis, and the activation of drug resistance pathways3,18,19,20. Mutations within histones or chromatin remodeling complexes can alter cell phenotype, contributing to neoplastic transformation. The accumulation of epigenetic alterations, such as histone methylation, is a key factor in activating drug resistance pathways21,22,23. Mutations within the SWI/SNF complex, which consists of 15 subunits encoded by 29 genes, impact over 20% of human cancers across many tumor types. In the past decade, epigenetic-based drug strategies (epi-drugs) have been developed, and many drugs targeting cancer with abnormal histone modification patterns have been approved by the US Food and Drug Administration (FDA)23.
The analysis of histone proteins is quite challenging, making it a demanding area of study. Recently, mass spectrometry methods have become the preferred way for characterizing and studying histone proteoforms. However, the drastic extraction conditions, in combination with the poor solubility of some variants and the high sequence homology, make their analysis challenging.
In the past decade, several mass spectrometry-based methods have been developed for histone profiling and investigation24,25,26,27. In this study, we present a cost-effective and reliable method to create a two-dimensional map of histone proteoforms, which can enhance the chances of discovering new post-translational modifications.
2D TAU electrophoresis combines separation in acidic conditions with SDS PAGE. In the first dimension, the medium's acidic condition gives a net positive charge to basic proteins. Since histones are very basic proteins and have a higher isoelectric point (pH) compared to the medium, they become positively charged and migrate under an electric field. In the second dimension, histones are denatured by SDS and separated based on their molecular weight. The resulting 2D map can be used as a tool to enrich specific proteoforms at single-spot resolution for mass spectrometry analysis and immunological studies28,29. After electrophoresis, 2D TAU spots can be destained, trypsin-digested, and analyzed by mass spectrometry. Alternatively, the 2D TAU map can be blotted onto a nitrocellulose/PVDF filter and investigated by immunostaining27.
In this context, the methods might be strategic to improve the identification and analysis of novel post-translational modifications correlated with metabolic rewiring30,31.
We have further improved the original method by incorporating a sonication step, which is helpful for DNA degradation and improving protein solubility. Additionally, we have prolonged the incubation step for histone extraction, in order to obtain a protein sample with high purity. Moreover, in our protocol, we ran the first dimension at low voltage for a longer duration to achieve a better resolution. The resulting gel was used for investigating canonical histones PTMs and characterizing novel modifications such as glycation10,32.
Overall, we have developed a method for extracting and purifying histones, followed by the resolution of histone proteoforms using a two-dimensional map. The work also includes a procedure for in-gel digestion for mass spectrometry analysis and the steps for two-dimensional western blot analysis.
Protocol
1. Histone extraction
- Perform histone extraction from at least 1 x 106 cells, which are 80% confluent. For this experiment, we used MCF-10A cell lines from ATCC. After plating, wash cells thrice with 1 mL of phosphate-buffered saline (PBS) 1x. Perform all the steps on ice.
- Prepare a cell lysis buffer containing 10 mM Tris-HCl pH 8.0, 1 mM KCl, and 1.5 mM MgCl2. Add 1 mM dithioerythritol and protease and phosphatase inhibitors just before use.
- Add 800 µL of cell lysis buffer to the cell-containing Petri dish, scrape, and collect the cells in a 1.5 mL tube, then incubate on a rotator at 300 rpm, 4 °C for 1 h.
- Centrifugate at 10,000 x g, 4 °C, for 10 min in a precooled microcentrifuge with a rotator for 1.5 mL tubes.
- Wash the pellet with 400 µL of cell lysis buffer and centrifuge at 10,000 x g, 4 °C, for 10 min. Discard the supernatant. Repeat the washing step 3x.
NOTE: Use a precooled microcentrifuge (4 °C) with a rotator for 1.5 ml tubes for all the steps. Handle the pellet carefully to avoid loss of material. - Resuspend the pellet in 640 µL of 0.2 M H2SO4 and incubate on a rotator at 300 rpm, at 4 °C, for 16 h.
CAUTION: Sulfuric acid is a hazardous chemical; please use it under a chemical fume hood. - Centrifuge at 16,000 x g, 4 °C, for 10 min. Transfer the supernatant to a fresh tube and store it until the following steps. Discard the nuclear debris.
- Precipitate histones by adding 212 µL of Trichloroacetic acid, drop by drop (the final concentration of TCA is 33%). Mix the solution by inverting the tube 10x. Incubate on ice, in the dark, for 16 h.
CAUTION: TCA is a hazardous chemical; use under a chemical fume hood. - Pellet histones by spinning in a precooled centrifuge at 16,000 x g for 10 min at 4 °C. Carefully remove the supernatant and wash the pellet 3x with 200 µL of cold acetone.
- Resuspend the pellet with cold acetone, then sonicate high power 2x with 3 cycles of 30 s on/off, alternating with a 10 min pause on ice.
NOTE: The procedure was done using an ultrasonicator. This step is essential to eliminate histone-wrapped DNA. - Centrifuge at 16,000 x g for 10 min, discard acetone, and resuspend the sample in 100 µL of double-distilled water. Determine protein concentration using the Bradford assay method, according to the manufacturer's instructions33.
NOTE: After centrifugation, the pellet sometimes appears white or translucent. A white and opaque precipitate could indicate the presence of salt contaminants that might impair the 2D separation. If so, repeat the acetone wash steps until the pellet becomes translucid. - Lyophilize 70 µg of histones, according to the manufacturer's instructions. Set the lyophilizer at 30 °C for 60 min. Check the tube volume and reiterate the step at 30 °C until the sample is completely lyophilized (generally, 90 min is necessary to completely lyophilize the sample). After 60 min, check the volume every 10 min since the process is sample-dependent.
NOTE: If the amount of histone proteins is low, start from 2 x 106 cells. In this case, use the same volume of reagents used for 1 x 106 cells. Check the purity of all the chemicals used during the procedure; all the reagents must be HPLC grade.
2. Mini 2D gel triton-acid-urea (TAU) electrophoresis
- Prepare a mini-TAU gel for a mini vertical polyacrylamide gel electrophoresis system as described below.
- Prepare separating gel: 6 M Urea, 15% acrylamide/bisacrylamide (29:1), 5% glacial acid acetic, 0.25% Triton X-100 (v/v), 0.4% TEMED (v/v), 0.1% APS (p/v). To prepare the solution, dissolve urea in acrylamide/bis and 1 mL of ultrapure water with gentle agitation at room temperature; add the other components and adjust the volume. For a mini-TAU gel, prepare 10 mL of solution. Fill the preset gel plates with the solution, avoiding the formation of air bubbles just 1.5 cm from the top. Cover the solution with 1 mL of isopropanol to level out the gel.
- Prepare stacking gel: 6 M Urea, 6% acrylamide/bisacrylamide (29:1), 5% glacial acid acetic, 0.25% Triton X-100 (v/v), 0.4% TEMED (v/v), 0.1% APS (p/v). To prepare the solution, dissolve urea in acrylamide/bis and 1 mL of ultrapure water; add the other components and adjust to volume. Fill gel plates with stacking solution, place a 10-well gel comb between plates, and insert it until the gel solution comes halfway up the teeth of the comb. Allow the gel to polymerize 16 h at 4 °C.
NOTE: Carefully insert the comb to ensure the regular formation of wells.
- Prepare 100 µL of TAU sample buffer by dissolving 6 M Urea, 5% glacial acid acetic, and double-distilled water. Dissolve the lyophilized sample prepared in 30 µL of TAU sample buffer.
- Fill the mini cassette with TAU running buffer, 5% acetic acid final concentration. Rinse the wells of the TAU-gel with a syringe filled with running buffer and load the sample in the well using a micropipette.
- Connect the electrophoretic chamber to the power supply, with the positive (red) electrode placed at the bottom. Pay attention, when IP > pH, proteins are positively charged, so the migration is from positive to negative electrode. Run the gel first at 30 V for 70 min, then at the constant voltage of 25 V for 17 h.
NOTE: During this time, occasionally switch off the power supply and rinse the gel wells using a disposable syringe. Pay attention to removing any bubbles from the bottom surface of the gel using a syringe to avoid the loss of sample and the presence of a smearing in the resolved proteins. - Remove the gel from the gel plates and cut the line of interest with a clean scalpel. Carefully handle the gel to avoid gel breakage. Since the line of interest is colorless, load the sample in well number 1 to exactly identify and cut the lane of interest.
NOTE: To check the separation on mini-TAU gel, prepare and run the sample in duplicate. One of these gels can be stained using Coomassie Brilliant Blue. The pattern of separation is reported in Figure 1A. - Wash the lane with 15 mL of 0.5 M Tris-HCl pH 8.8 for 15 min.
- Prepare 15 mL of equilibration buffer 1 using 6 M Urea, 0.5 M Tris-HCl, pH 6.8, 30% glycerol, 20% SDS, and 2% Dithioerythritol. Prepare 15 mL of equilibration buffer 2, using 6 M Urea, 0.5 M Tris-HCl, pH 6.8, 30% glycerol, 20% SDS, and 2% Iodoacetamide. Prepare the equilibration buffer 1 and 2 just before use; store the equilibration buffer 2 in the dark.
- Incubate the gel lane, under gentle agitation, in 10 mL of equilibration buffer 1 for 15 min, then decant. Incubate the gel lane, under gentle agitation, with 10 mL of equilibration buffer 2 for 15 min in the dark, then decant.
- Prepare a resolving SDS 15% acrylamide gel by following Table 1. Set up the gel plate and fill it with the resolving gel solution. Fill the solution until it is 1.5 cm from the end of the long gel plate. Put 1 mL of isopropanol on the surface to level the gel and allow it to polymerize.
- After gel polymerization, decant the isopropanol and dry the well with 3MM Chr Cellulose Chromatography Paper. Insert the resulting lane from step 8 into the gel well.
- Seal the lane to the resolving gel with a sealing solution of 0.5% low melting point agarose in 1x Tris/glycine/SDS buffer and 5 µL of 0.003% bromophenol blue. Allow agarose gel polymerization for 30 min.
NOTE: Avoid the formation of air bubbles during agarose sealing to prevent gel smearing. - Put the gel plates in the cassette and fill with 1x Tris/glycine/SDS buffer. Run the gel at 120 V until the bromophenol blue dye reaches the bottom. Gently remove the gel from the plates, discard the excess of agarose, and silver stain the gel using a procedure compatible with mass spectrometry or blot for immunological assay.
3. Silver staining 32
- Prepare the following reagents – Fixative: 50% methanol, 5% acetic acid, 45% deionized water; Wash solution: 50% methanol, 50% deionized water; Sensitizer: 0.02% Na2S2O3 5H2O in deionized water; Silver reagent: 0.1% silver nitrate in cold (4°C) deionized water; Developer: 0.04% Formalin and 2% Na2CO3, Formaldehyde (37%) (add just before use) 0.4 mL deionized water and fill up to 1000 mL; Stop-reagent: 5% acetic acid; Gel storage solution: 1% acetic acid.
NOTE: Since chemicals used for staining are hazardous, perform all the steps under a chemical fume hood. - Follow the procedure in Table 2 with the following consideration. Perform all incubations with gentle agitation. Prepare the stop reagent before starting the developing step; this will ensure gel development with the opportune intensity. During the developing step, spots will appear as yellow signals that will become light brown at the end of the incubation. The intensity of the different spots will depend on the amount of histone proteoforms. The gel is ready for spot excision and in-gel digestion (Figure 1B)
4. In-gel digestion
- Prepare the following solutions: 50 mM ammonium bicarbonate (NH4HCO3)/50% (v/v) acetonitrile; 10 mM dithiothreitol (DTT) in 50 mM NH4HCO3; 55 mM iodoacetamide (IAA) in 50 mM NH4HCO3; 0.1% TFA; 50 mM NH4HCO3, pH 8; HPLC grade Acetonitrile; 1 mL silicon tubes.
NOTE: To circumvent human keratin contamination, from hair or skin, use clean gloves and work in a clean area. - Excise histone spots of interest from the stained gel using a clean scalpel and place them into a fresh silicon tube (1.5 mL). Take care to excise a single spot and put it in a single tube to avoid isoform cross-contamination.
- Add to each gel spot 250 µL of 50 mM NH4HCO3/50% acetonitrile and incubate under gentle agitation in a thermomixer at room temperature. Decant the solution and repeat this step 3x. Gel pieces will appear transparent.
- Dehydrate gel spots by adding acetonitrile (200 µL). At this stage the gel pieces will take an opaque-white color.
- Decant acetonitrile and let air-dry for 10 min. The gel piece will appear dried in the tube. Reduction and alkylation are not essential since histone proteins have been reduced and alkylated during the equilibration step.
- Rehydrate gel spots in 20 µL of trypsin solution (0.1 mM HCl) or a volume sufficient to cover the gel pieces. Place the tubes in the thermomixer and incubate, under gentle agitation, at 37 °C for a minimum of 4 h until 16 h.
- Transfer the supernatant containing the tryptic peptides to a fresh tube. Add 50 µL of extraction solution (60% acetonitrile, 1% TFA) to the gel pieces and sonicate 2x in an ultrasonic water bath for 10 min at 4 °C.
- Transfer the supernatant, containing additional tryptic peptides, to the fresh tube. Use a lyophilizer to dry the pooled extracted peptides. Set the lyophilizer at 30 °C and dry the solution; a lyophilized pellet will appear in the tube.
NOTE: This step is volume-dependent; generally, 1 h is enough, but check the volume every 10 min until the sample is completely dried. - Add 15 µL of 0.1 % TFA to each tube and gently incubate in a thermomixer at 25 °C. Transfer the supernatant to the vial for LC-MS/MS analysis.
5. 2DTAU western blot
- Prepare the transfer buffer, 250 mM Tris base, 92 mM glycine, and 10% methanol using double-distilled water.
- Cut the nitrocellulose membrane and the filter paper to the dimensions of the gel.
- Equilibrate the gel and soak the membrane, filter paper, and filter papers in the transfer buffer for 15 min. Use either Nitrocellulose or PVDF membranes. If using PVDF (Polyvinylidene difluoride) membranes, activate them by soaking in 100% MetOH.
- Prepare the gel sandwich in the cassette for a Semi-dry transfer as follows: Negative electrode/Filter-Paper/Gel/Nitrocellulose-membrane/Filter-paper/Positive electrode. Remove any air bubbles between gel and membrane to ensure good results.
- Place the cassette in the Semi-dry unit and run the blot at 25 V for 30 min. At the end, disassemble the blotting sandwich and store the membrane for the immunological assay. Check the transfer efficiency with Ponceau staining. Mark the membrane with a sign at the top right to preserve the right filter orientation.
- Incubate with primary and secondary antibodies depending on the experiment (Figure 2).
Representative Results
In Figure 1A, it is possible to observe how histone proteins migrate in a 1D gel. This is an important preliminary step to understand the running time of the first dimension. Once the right time of separation is determined, run the second dimension. The 2D map of histone proteoforms shows a distinctive topographic pattern (Figure 1B). Each spot in Figure 1B represents a specific histone type. Observe the exact region of the gel where each histone proteoform can be found. The mass spectrometry identification for each histone proteoform is reported in Perri et al.32. Therefore, each spot in this condition is associated with a specific histone type. The presence of additional spots, alongside the canonical ones, may indicate the existence of novel modifications. Gel images can be acquired using densitometry, and the resulting image can be analyzed using image analysis software.
The reliability of the 2D TAU map is dependent on the spot's rotundity. If the run has been performed correctly, the spots will appear as distinct signals and no horizontal and vertical streaking will be visible. Conversely, impurities in the sample, solutions, and agarose overlay might cause streaks and/or stripes in the gel. The quality of the gel will impact the image digitalization and the following quantitative and qualitative analysis. Moreover, it might influence the success of Western Blot analysis and MS investigations. To verify the reliability of the method, we probed the nitrocellulose membrane of resolved histone proteoforms with the primary antibody Acetylated-Lysine (Ac-K2-100) Rabbit mAb32 (Figure 2). An anti-rabbit HRP secondary antibody was used to detect histone signals. As expected, using a densitometer, it is possible to quantify the peculiar post-translational modification on each histone variant present in the blot.
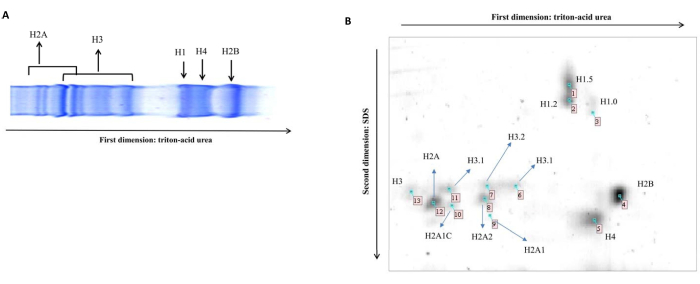
Figure 1: 1D and 2D TAUgel. (A) Representative image of histones topographic pattern obtained using 1D TAUgel. (B) Representative image of histones topographic pattern obtained using 2D TAUgel. Data identifications are reported by Perri et al. 32. As expected, each spot represents a specific histone proteoform. Please click here to view a larger version of this figure.

Figure 2: Representative 2DTAU Western blot. In the image, it is possible to observe the level of lysine acetylation in each histone proteoform. Please click here to view a larger version of this figure.
| Reagents | Concentration: 15% |
| 40% Acrylamide/Bis | 3.75 |
| 1.5 M Tris pH 8.8 | 2.5 mL |
| MilliQ water | 3.6 mL |
| 10% SDS | 100 µL |
| 10% APS | 50 µL |
| TEMED | 5 µL |
Table 1: Resolving gel solution composition.
| Step | Time |
| Fixing 50% methanol / 5% acetic acid | 1 h |
| Wash 30% ethanol | 2 x 20 min |
| Wash deionized water | 1 x 20 min |
| Sensitizer Thiosulfate reagent | 1 min |
| Wash deionized water | 3 x 20 s |
| Silver Silver nitrate | 30 min |
| Wash deionized water | 3 x 1 min |
| Development Developer | 25-30 min Note: Develop the gel until the spots are visible (light brown) |
| stopping solution | 1 x 10 min |
| Wash deionized water | 2 x 5 min |
| Wash deionized water | 1 h |
Table 2: Silver staining method.
Discussion
In the past decade, cancer patient therapy has been revolutionized by identifying various molecular alterations that drive cancer development and progression. The most significant advancement in modern oncology is the development of therapy based on comprehensive molecular analysis. Technological advances in genomics and proteomics have played a crucial role in profiling specific biomarkers for early detection, surveillance, prognosis, and drug monitoring. Proteomics is defined as the analysis of the complete protein complement of a cell, tissue, or organism under a particular condition, and it also includes the study of protein interactions, post-translational modifications, and localization34. A specific area of proteomics is epi-proteomics, which involves the systematic study of histone post-translational modifications. These have been linked to the development and progression of cancer35. The paper outlines a method for identifying specific histone isoforms that contribute to the cellular phenotype.
The critical step of the method is extracting histones because of their poor solubility and high basicity. Its major limitations are the complexity of the 2D map and the low number of variants, which might make the MS/MS analysis difficult.
Overall, the major benefit of this approach is that it can be carried out in any laboratory, and a mini gel apparatus can be owned with the appropriate precautions. In this context, we are confident that this method might aid a large number of scientists, such as those who cannot benefit from a large facility in their institution, to identify novel epi-markers, aiming at improving the knowledge about the epi-proteome. Furthermore, this method can be combined with mass spectrometry, which can be outsourced as a service, to discover novel proteoforms. It can also be used with immunological assays to confirm the changes in a specific post-translational modification for which a commercial antibody is available.
The method has been used to study the typical changes in histone modifications related to the development of breast cancer28 and to examine the modifications of H1 in mouse embryonic stem cells36. Additionally, the method has been successfully employed to measure the level of histone carbonylation37 in rapidly growing fibroblasts and to explore new histone modifications, such as histone glycation and histone MARylation38.
From our perspective, we are confident that this method can be successfully applied for profiling novel histone PTMs, particularly those associated with the abnormal activation of specific metabolic pathways, such as acetylation, malonylation, methylation, lipidation, lactylation, butylation, and others. Ultimately, the method could also serve as a micro-preparative tool for isolating individual proteoforms and may facilitate the analysis of metabolism-linked PTMs, thereby enhancing our understanding of this crucial area of research.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The work was supported by PRIN2022, DS, CF, and AC were supported by PRIN2022, and MLC was supported by PhD program in Molecular Oncology, UMG PhD program school. The work was also supported by Project PNRR INSIDE CUP: B83C22003920001
Materials
| 1,4-Dithioerythritol | SIGMA- ALDRICH | D8255 | Reagent for maintaining −SH groups in the reduced state; quantitatively reduces disulfides. |
| 1,5 M Tris-HCL buffer, pH 8.8 | BIO-RAD | 161-0798 | Resolving Gel Buffer |
| 10x Tris/Glycine/SDS | BIO-RAD | 1610772 | Use this premixed 10x Tris/glycine/SDS running buffer to separate protein samples by SDS-PAGE. |
| 2-Propanol | SIGMA-ALDRICH | 33539 | 2-Propanol (Isopropanol) is a secondary alcohol. It has been tested as a substitute to fuel for use in various fuel cells. CuO powder dissolved in 2-propanol has been used for the laser ablation assisted synthesis of Cu colloids. Suspension of 2-propanol with Zn(NO3)2 has been employed for the fabrication of titanium dioxide-coated stainless steel (P25-TiO2/SS) photoanode coated with uniformly thick layer of P25-TiO2.This photoanode was used for the electrochemical photocatalytic (ECPC) degradation process. |
| 30% Acrylamide/Bis Solution | BIO-RAD | 1610158 | 2.6% Crosslinker. Electrophoresis purity reagent |
| Acetic Acid Glacial | Carlo Erba Reagennts | 401392 | Acetic acid is an important chemical reagent with many industrial applicatios. |
| Acetone | Panreac Applichem | 211007.1214 | Acetone is a solvent, renowned for its versatility in various laboratory, manufacturing, and cleaning applications. |
| Acetonitile | VWR | 83640320 | for HPLC LC-MS grade |
| Agarose | Invitrogen | 15510-027 | Agarose is a heteropolysaccharide frequently used in molecular biology. |
| Ammonium bicarbonate | SIGMA-ALDRICH | A6141 | minimum 99,0% |
| Ammonium persulfate | SIGMA-ALDRICH | A3678 | For molecular Biology, For electrophoresis, ≥98% |
| Bioruptor plus | Diagenode | B01020001 – B01020002- B01020003 | sonicator |
| Dithiothreitol | SIGMA- ALDRICH | D9163-5G | Reducing agent used to reduce disulfide bonds in proteins. |
| Dodeca Silver Stain Kit | BIO-RAD | 161-0480 | The Dodeca silver stain kit is easy-to-use for the detection of nonagram levels of proteins in polyacrylamide gels. |
| Glycerol | GE – Healthcare Life Sciences | 171325010L | Glycerol is a simple triol compound, it is involved to aid in casting gradient gels, protein stabilizer and storage buffer component. |
| Halt Phosphatase Inhibitor | Thermo SCIENTIFIC | 78428 | Thermo Scientific Halt Phosphatase Inhibitor Cocktail preserves the phosphorylation state of proteins during and after cell lysis or tissue protein extraction Single- Use Cocktail (100X). |
| Halt Protease Inhibitor | Thermo SCIENTIFIC | 78430 | Thermo Scientific Halt Protease Inhibitor Cocktail (100X) are ready-to-use concentrated stock solutions of protease inhibitors for addition to samples to prevent proteolytic degradation during cell lysis and protein extraction. Single- Use Cocktail (100X). |
| Hydrochloric acid | SIGMA-ALDRICH | H1758 | BioReagent, for molecular biology |
| Iodoacetamide | SIGMA-ALDRICH | I6125 | ≥99% (NMR), crystalline |
| KCl | SIGMA-ALDRICH | P9541 | Potassium chloride, KCl, is generally used in laboratory routines. Its use as a storage buffer for pH electrodes and as a reference solution for conductivity measurements is well established. |
| MCF10 cell line | atcc | CRL-10317 | epithelial cell line that was isolated in 1984 from the mammary gland of a White, 36-year-old female with fibrocystic breasts. This cell line was deposited by the Michigan Cancer Foundation. |
| MgCl2 | SIGMA-ALDRICH | 208337 | Magnesium chloride is a colorless crystalline solid. |
| N,N,N',N'-Tetramethylethylene-diamine | SIGMA-ALDRICH | T9281 | For Electrophoresis, approx. 99% |
| Phosphate saline buffer 1X | Corning | 15373631 | 1 X PBS (phosphate buffered saline) is a buffered balanced salt solution used for a variety biological and cell culture applications, such as washing cells before dissociation, transporting cells or tissue, diluting cells for counting, and preparing reagents. |
| Potassium hexacyanoferrate | SIGMA-ALDRICH | 60299 | Electron acceptor, employed in systems involving electron transport, |
| SDS Solution 20% (w/v) | BIO-RAD | 161-0418 | Sodium dodecyl sulfate. Electrophoresis purity reagent |
| Sodium thiosulfate, ReagentPlus 99% | SIGMA- ALDRICH | 21,726-3 | Sodium thiosulfate, ReagentPlus 99% |
| Sulfuric acid | SIGMA-ALDRICH | 339741 | Sulfuric Acid, Reagent, is an extremely corrosive acid that comes as yellowy slightly viscous liquid. It is soluble in water and is a diprotic acid. It has very strong corrosive, dehydrating and oxidizing properties as well as being hygroscopic. |
| Trichloroacetic acid | SIGMA-ALDRICH | T6399-5G | Trichloroacetic acid (TCA) is used as a reagent for the precipitation of proteins1,2 and nucleic acids3. |
| Trifluoroacetic acid | Riedel-de Haën | 34957 | Trifluoroacetic acid |
| Triton X-100 | SIGMA- ALDRICH | T9284-1L | Triton X-100 is a nonionic polyoxyethylene surfactant that is most frequently used to extract and solubilize proteins. |
| Trypsin from porcine pancreas | SIGMA-ALDRICH | T6567 | Proteomics Grade, BioReagent, Dimethylated |
| UREA | SIGMA-ALDRICH | 33247 | Urea is a chaotropic agent and is used for protein denaturation. It disturbs the hydrogen bonds in the secondary, tertiary and quaternary structure of proteins. It can also disturb hydrogen bonding present in DNA secondary structure |
References
- Strahl, B. D., Allis, C. D. The language of covalent histone modifications. Nature. 403 (6765), 41-45 (2000).
- Cutter, A. R., Hayes, J. J. A brief review of nucleosome structure. FEBS Lett. 589 (20 Pt A), 2914-2922 (2015).
- Millán-Zambrano, G., Burton, A., Bannister, A. J., Schneider, R. Histone post-translational modifications – cause and consequence of genome function. Nat Rev Genet. 23 (9), 563-580 (2022).
- Sun, L. et al. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 13 (12), 877-919 (2022).
- Kinnaird, A. et al. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 16 (11), 694-707 (2016).
- Díaz-Hirashi, Z. et al. Metabolic reprogramming and signaling to chromatin modifications in tumorigenesis. Adv Exp Med Biol. 1219, 225-241 (2020).
- Panatta, E. et al. Metabolic regulation by p53 prevents R-loop-associated genomic instability. Cell Rep. 41 (5), 111568 (2022).
- Scumaci, D., Zheng, Q. Epigenetic meets metabolism: novel vulnerabilities to fight cancer. Cell Commun Signal. 21 (1), 249 (2023).
- Chiaradonna, F., Scumaci, D. Cancer metabolism as a new real target in tumor therapy. Cells. 10 (6), 1393 (2021).
- Scumaci, D. et al. DJ-1 proteoforms in breast cancer cells: The escape of metabolic epigenetic misregulation. Cells. 9 (9), 1968 (2020).
- Olivo, E. et al. Moving beyond the tip of the iceberg: DJ-1 implications in cancer metabolism. Cells. 11 (9), 1432 (2022).
- Zheng, Q., Maksimovic, I., Upad, A., David, Y. Non-enzymatic covalent modifications: a new link between metabolism and epigenetics. Protein Cell. 11 (6), 401-416 (2020).
- Maksimovic, I., David, Y. Non-enzymatic covalent modifications as a new chapter in the histone code. Trends Biochem Sci. 46 (9), 718-730 (2021).
- Lowe, B. R. et al. Histone H3 mutations: an updated view of their role in chromatin deregulation and cancer. Cancers. 11 (5), 660 (2019).
- Karsli-Ceppioglu, S. et at. The epigenetic landscape of promoter genome-wide analysis in breast cancer. Sci Rep. 7 (1), 6597 (2017).
- Yang, C. et al. Histone methyltransferase and drug resistance in cancers. J Exp Clin Cancer Res. 39 (1),173 (2020).
- Vardabasso, C. et al. Histone variants: emerging players in cancer biology. Cell Mol Life Sci. 71 (3), 379-404 (2014).
- Ilango, S., Paital, B., Jayachandran, P., Padma, P. R., Nirmaladevi, R. Epigenetic alterations in cancer. Front Biosci. 25 (6), 1058-1109 (2020).
- Liu, Y. et al. Histone H2AX promotes metastatic progression by preserving glycolysis via hexokinase-2. Sci Rep. 12 (1), 3758 (2022).
- Nowsheen, S. et al. ZNF506-dependent positive feedback loop regulates H2AX signaling after DNA damage. Nat Commun. 9 (1), 2736 (2018).
- Ribeiro-Silva, C., Vermeulen, W., Lans, H. SWI/SNF: Complex complexes in genome stability and cancer. DNA Repair. 77, 87-95 (2019).
- Yang, Y., Wang, Y. Role of epigenetic regulation in plasticity of tumor immune microenvironment. Front Immunol. 12, 640369 (2021).
- Miranda Furtado, C. L. et al. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 14 (12), 1164-1176 (2019).
- Thomas, S. P. et al. A practical guide for analysis of histone post-translational modifications by mass spectrometry: best practices and pitfalls. Methods. 184, 53-60 (2020).
- Noberini, R. Robusti, G. Bonaldi, T. Mass spectrometry-based characterization of histones in clinical samples: applications, progress, and challenges. FEBS J. 289 (5), 1191-1213 (2022).
- Kwak, H. G., Dohmae, N. Proteomic characterization of histone variants in the mouse testis by mass spectrometry-based top-down analysis. Biosci Trends. 10,357-364 (2016).
- Sidoli, S., Simithy, J., Karch, K.R., Kulej, K., Garcia, B.A. Low resolution data-independent acquisition in an LTQ-Orbitrap allows for simplified and fully untargeted analysis of histone modifications. Anal Chem. 87, 11448-11454 (2015).
- Shechter, D., Dormann, H. L., Allis, C. D., Hake, S. B. Extraction, purification and analysis of histones. Nat Protoc. 2 (6), 1445-1457 (2007).
- Basak, S. K., Ladisch, M. R. Correlation of electrophoretic mobilities of proteins and peptides with their physicochemical properties. Anal Biochem. 226 (1), 51-58 (1995).
- Nuccio, A. G., Bui, M., Dalal, Y., Nita-Lazar, A. Mass spectrometry-based methodology for identification of native histone variant modifications from mammalian tissues and solid tumors. Methods Enzymol. 586, 275-290 (2017).
- Zhang, S., Roche, K., Nasheuer, H. P., Lowndes, N. F. Modification of histones by sugar β-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J Biol Chem. 286 (43), 37483-37495 (2011).
- Perri, A. M. et al. Histone proteomics reveals novel post-translational modifications in breast cancer. Aging. 11, 11722-11755 (2019).
- Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72, 248-254 (1976).
- Omenn, G. S. et al. Research on the human proteome reaches a major milestone: >90% of predicted human proteins now credibly detected, according to the HUPO human proteome project. J Proteome Res. 19 (12), 4735-4746 (2020).
- Noberini, R., Robusti, G., Bonaldi, T. Mass spectrometry-based characterization of histones in clinical samples: applications, progress, and challenges. FEBS J. 289 (5),1191-1213 (2022).
- Starkova, T. Y. et al. The profile of post-translational modifications of histone H1 in chromatin of mouse embryonic stem cells. Acta Naturae. 11 (2), 82-91 (2019).
- García-Giménez, J. L. et al. Histone carbonylation occurs in proliferating cells. Free Radic Biol Med. 52 (8), 1453-1464 (2012).
- García-Saura, A. G., Schüler, H. PARP10 multi-site auto- and histone MARylation visualized by acid-urea gel electrophoresis. Cells. 10, 654 (2021).

.