Automatically Generated
Nuclear Transport Assays in Permeabilized Mouse Cortical Neurons
Summary
We have developed a reliable method of selective plasma membrane permeabilization of primary mouse cortical neurons for high content automated analysis of neuronal nucleocytoplasmic transport.
Abstract
Disruption of nucleocytoplasmic transport is increasingly implicated in the pathogenesis of neurodegenerative diseases. Moreover, there is a growing recognition of cell-specific differences in nuclear pore complex structure, prompting a need to adapt nuclear transport methods for use in neurons. Permeabilized cell assays, in which the plasma membrane is selectively perforated by digitonin, are widely used to study passive and active nuclear transport in immortalized cell lines but have not been applied to neuronal cultures. In our initial attempts, we observed the rapid loss of nuclear membrane integrity in primary mouse cortical neurons exposed to even low concentrations of digitonin. We hypothesized that neuronal nuclear membranes may be uniquely vulnerable to the loss of cytoplasmic support. After testing multiple approaches to improve nuclear stability, we observed optimal nuclear integrity following hypotonic lysis in the presence of a concentrated bovine serum albumin cushion. Neuronal nuclei prepared by this approach reliably import recombinant fluorescent cargo in an energy-dependent manner, facilitating analysis of nuclear import by high content microscopy with automated analysis. We anticipate that this method will be broadly applicable to studies of passive and active nuclear transport in primary neurons.
Introduction
Disruption of nucleocytoplasmic transport, the regulated trafficking of proteins and RNA between the nucleus and cytoplasm, is increasingly implicated in the pathogenesis of neurodegenerative diseases (recently reviewed1). We and others have reported structural and functional disruption of the nucleocytoplasmic transport apparatus in postmortem tissue and animal models of C9orf72 amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), Alzheimer’s disease, and Huntington's disease2,3,4,5. The mechanisms and functional consequences of nucleocytoplasmic transport disruption for neurodegeneration, and approaches for therapeutic rescue, are areas of ongoing investigation.
Nuclear pores are large transmembrane complexes of ~30 nucleoporin proteins that permit diffusion of small molecules across the nuclear membrane but increasingly restrict the passage of cargoes >40 kD via a permeability barrier of phenylalanine-glycine (FG)-rich nucleoporins in the central channel6. Larger cargoes containing nuclear localization signal (NLS) or nuclear export signal (NES) sequences undergo active, receptor-mediated transport across the pore via nuclear transport receptors (importins and exportins) and a steep gradient of the small GTPase Ran across the nuclear membrane (recently reviewed7). A wide array of methods has been developed to analyze nuclear transport dynamics in cultured cells, including the trafficking of endogenous cargoes and tagged reporter constructs that serve as substrates for the major subclasses of transport receptors. Such approaches have been readily adapted to neurons2,5,8 and provide a readout of nuclear transport perturbations in the context of an intact, living cell. However, in live cell assays, the ability to directly manipulate nuclear transport reactions or investigate them in isolation from other cellular processes is limited.
In permeabilized cell assays, the plasma membrane is selectively perforated, and the cytoplasm is released, leaving the nuclear envelope and nuclear pore complexes intact and able to perform either passive or energy-dependent bidirectional transport9,10. Such transport reactions can be readily reconstituted by adding whole-cell lysates, cytoplasmic fractions, or purified recombinant nuclear transport proteins and their cargos. Thus, permeabilized cell assays permit a broad range of biochemical or biophysical investigations, including delivery of recombinant or synthetic proteins and RNAs relevant to the study of neurodegenerative diseases.
Given reports of cell-specific differences in nuclear pore complex structure and transport dynamics11,12, we aimed to adapt the permeabilized cell assay for use in primary neuronal cultures. Although widely used to analyze nuclear transport in immortalized cell lines, despite exhaustive literature search, we found no published reports of neuronal plasma membrane permeabilization that verified preservation of nuclear membrane integrity. Most protocols rely on digitonin, a detergent that targets the unique cholesterol composition of the plasma membrane, to perforate the nuclear membrane while leaving the nuclear membrane intact13. Our initial attempts using digitonin in primary mouse cortical neurons showed immediate loss of nuclear membrane integrity, evidenced by diffusion of a 70 kD fluorescent dextran into the nucleus. We hypothesized that nuclear envelope rupture might be caused by mechanical perturbation from loss of cytoplasmic support, and tested multiple methods of optimization, including molecular crowding, cytoskeletal stabilization, and alternate methods of cell lysis. Here, we detail a method of rapid hypotonic permeabilization using a concentrated bovine serum albumin (BSA) cushion to protect neuronal nuclei and facilitate downstream analysis of neuronal nuclear import. We recently used this method to evaluate the mechanism of dipeptide repeat protein disruption in C9orf72-ALS/FTD14 and anticipate that it will be broadly applicable to future studies of passive and active nuclear transport in primary neurons.
Protocol
First, the protocol describes the generation of primary neuronal cultures (step 1) and preparation of materials for the transport assay (step 2), followed by the transport assay itself (steps 3-4) and image acquisition and analysis (step 5). All methods described here were approved by the Animal Care and Use Committee (ACUC) of Johns Hopkins University.
1. Primary mouse cortical neuron cultures
- Prepare the stock solutions.
- Dissection buffer: Combine 50 mL of 10x HBSS, 15 mL of 1 M glucose (in dH2O), 5 mL of 1 M HEPES, 5 mL of 100 mM sodium pyruvate, and 5 mL of 100x penicillin-streptomycin. Bring volume to 500 mL with dH2O. Sterile filter and store at 4 °C.
- Papain: Dilute papain to 30 mg/mL with dissection buffer. Store at 4 °C.
- DNase: Dilute DNase I to 10 mg/mL with dissection buffer. Aliquot and store at -20 °C.
- Plating medium: Add 5% fetal bovine serum (FBS), 2% B-27, 1% Glutamax, and 1% penicillin-streptomycin to neurobasal medium. Sterile filter and store at 4 °C.
- Feeding medium: Add 2% B-27, 1% Glutamax, and 1% penicillin-streptomycin to Neurobasal medium. Store at 4 °C.
- Coat the plates.
- Coat the optical glass-bottom 96-well plates with poly-D-lysine (20 µg/mL) and laminin (1:100) in sterile dH2O. Place the plate in an incubator overnight.
- On the day of dissection, rinse the wells three times with sterile dH2O and replace with Neurobasal medium. Equilibrate in an incubator for at least 30 min.
- Dissect and digest the cortices.
NOTE: Carry out dissection on a clean bench using autoclaved tools to prevent culture contamination.- Euthanize the E15-16 C57BL/6J pregnant female by cervical dislocation. Spray abdomen with 70% ethanol. Work quickly. Use scissors and toothed forceps to make a midline abdominal incision, excise the uterus, and then transfer the embryos from the amniotic sac into a Petri dish. Decapitate the embryos with scissors and collect heads in a 10 cm Petri dish containing dissection buffer.
- Under a dissecting microscope, use fine forceps to remove the skull and peel away the meninges. Transfer whole brains to a fresh 10 cm Petri dish containing dissection buffer. Separate the hemispheres and remove the subcortical structures.
- Chop the cortices with a sterile razor blade and transfer them to a conical tube containing 5 mL of dissection buffer. Add 100 µL of papain (stock 30 mg/mL, final concentration 0.6 mg/mL) and 50 µL of DNase (stock 10 mg/mL, final concentration 0.1 mg/mL). Digest in a 37 °C water bath for 10 min.
NOTE: Digestion beyond 10 min is detrimental to cell survival. - Remove the supernatant and wash twice with 10 mL of plating media to stop digestion. Allow the tissue to settle by gravity in between each wash.
- Triturate gently in 2 mL plating media with P1000 pipette until large pieces of tissue disappear (approximately 20 times). Add another 8 mL of plating media to bring the volume to approximately 10 mL and pass the cell suspension through a 40 µm cell strainer.
- Plate and maintain neurons.
- Centrifuge at 200 x g for 5 min. Resuspend the cells in prewarmed plating medium. Count the cells and dilute to 500,000 cells/mL in the plating medium. Gently invert to mix and transfer to a sterile multichannel reservoir.
- Aspirate the neurobasal media from the wells and immediately add 100 µL of cells per well with a multichannel pipette (50,000 cells/well). Do not allow the coated wells to dry.
- After 24 h of dissection, change 50% of medium with the feeding medium. Continue 50% medium change every other day until day 5-7 to perform the in vitro assays.
2. Preparation of the nuclear transport assay components
- Transport buffer (TRB, adapted from15)
- To prepare 5x TRB, dissolve 100 mM HEPES, 550 mM KOAc, 10 mM Mg(OAc)2, 25 mM NaOAc, 2.5 mM EGTA, and 1.25 M sucrose in dH2O. Adjust the pH to 7.3 and store the buffer at 4 °C.
- On the day of the transport experiment, prepare 1x TRB-BSA. Dilute 5x TRB in dH2O, add EDTA-free protease inhibitor cocktail, 2 mM of DTT, and 50 mg/mL of BSA.
- Energy regeneration mix (ERM, adapted from9,16)
- To prepare 20x ERM, dissolve 2mM ATP lithium salt, 2 mM GTP lithium salt, 80 mM Creatine phosphate, and 400 U/mL Creatine kinase in 1x TRB.
- Freeze single-use aliquots at -20 °C.
- Preparation of whole cell extract (WCE, source of transport receptors and Ran cycle proteins).
NOTE: Cytoplasmic or whole cell extract (WCE) may be used, depending on the experimental goals. Here, the preparation of WCE is described as used in the initial neuron transport assays10.- Grow HEK293T cells to confluence in fifteen 150 mm culture dishes. Detach with trypsin-EDTA, resuspend in media containing 10% fetal bovine serum to inhibit trypsin, and pellet cells at 200 x g for 5 min. Resuspend the cells in 10 mL of ice-cold 1x TRB with freshly added protease inhibitor cocktail.
- Sonicate on ice, 3 x 10 pulses, using a hand-held probe sonicator. Clarify by centrifugation at 14,000 x g for 15 min at 4 °C.
- Measure the protein concentration (goal 8-10 mg/mL) and snap-freeze single-use aliquots in liquid nitrogen prior to storage at -80 °C.
- Recombinant nuclear import cargo
NOTE: The transport protocol in steps 3-5 may be adapted for any fluorescent nuclear transport cargo, to interrogate the active or passive nuclear transport pathway of interest. Here, the protocol describes the expression and nuclear import of Rango (Ran-regulated importin β cargo), which consists of the importin β-binding domain of importin α1 flanked by the fluorescent proteins CyPet and YPet14,17. Rango is a versatile sensor that can be used for FRET as well as nuclear import assays, where it functions as a direct importin β cargo.- Transform E. coli BL21(DE3) cells with the Rango plasmid (containing an N-terminal 6-His tag) by heat-shock as follows. Thaw a 50 µL aliquot of BL21(DE3) cells on ice. Combine with 100-200 ng plasmid. Mix by tapping the tube 4-5 times and then incubate on ice for 30 min before placing on a 42 °C plate for 40 s. Transfer immediately back to ice for another 2-5 min.
- Add 900 µL of SOC media at room temperature and incubate on a shaker at 37 °C for 1 h before spreading the cells on a prewarmed agar plate (1.5% agar, LB media, 100 µg/mL Ampicillin). Incubate the plate overnight at 37 °C. Store the plate at 4 °C for up to 1 week or use it immediately for protein production.
- Start the protein production by inoculating three 25 mL of starter cultures in 50 mL tubes with increasing number of colonies (~3-10) from the BL21(DE3) plate. After incubation overnight at 37 °C on a shaker, select the cultures with OD600 nm < 1.0 to inoculate a 1L culture in LB media using a 2.8 L baffle-free Fernbach flask. Grow at 37 °C until reaching OD600 nm = 0.1-0.3.
NOTE: The use of older BL21(DE3) LB agar plates or high density of starter cultures (OD600 nm >1.0) could reduce the yield and purity of the protein preparation. - Cool to room temperature (22-25 °C) by placing on ice. Induce protein expression with 0.3 mM IPTG (final concentration) and incubate on a shaker (80-120 rpm) at room temperature for 12-14 h.
- Collect the cells by centrifugation (6,000 x g, 15 min, 4 °C) and resuspend in 30 mL of ice-cold 10 mM imidazole in PBS (pH 7.4), supplemented with EDTA-free protease inhibitor cocktail. Lyse by two passes through an ice-cold French pressure cell and clarify the lysate by centrifugation (16,000 x g, 40 min, 4 °C).
- Incubate the lysate with high flow Nickel affinity agarose resin (30-60 min, 4 °C), using 1 mL of resin per 1 L of E. coli culture. Place the resin into the chromatography columns, wash with 10-20 volumes of ice-cold 10 mM imidazole/PBS (pH 7.4). Elute Rango with 25 mM imidazole/PBS increments (25-200 mM, pH 7.4), using a 5-fold bed volume in each step.
- Use SDS-PAGE to select and pool the batches with the highest purity.
- Prepare XB buffer by dissolving 10 mM HEPES, 100 mM KCl, 10 mM CaCl2, 1 mM MgCl2, and 50 mM sucrose in dH2O. Adjust the pH to 7.7.
- Dialyze the protein in XB buffer and concentrate by centrifugation (7500 x g, 4 °C) on an ultrafiltration device with a 30 kD molecular weight cutoff.
- Measure the protein concentration by the Bradford or other preferred method, and snap-freeze single-use aliquots in liquid nitrogen before storage at -80 °C.
3. Determination of the optimal plasma membrane permeabilization conditions
NOTE: Due to variations between each batch of neurons, optimize the permeabilization conditions for each batch of neurons. Perform this on the same day before performing nuclear transport assays.
- In a plastic 96-well staging plate, prepare a serial dilution of Tris-HCl pH 7.5 (to cause osmotic swelling), at 0 µM, 10 µM, 20 µM, and 40 µM in each of three BSA concentrations: 50 mg/mL, 100 mg/mL, and 150 mg/mL (for molecular crowding/mechanical support). Prewarm the solutions to 37 °C.
- Rinse the neurons once in prewarmed PBS. Remove the PBS and transfer Tris/BSA from the staging plate. Return to the incubator for 4 min.
- Remove from the incubator and place the culture plate on ice. Rinse 2 x 5 min in 1x TRB-BSA (as prepared in step 2.1.2).
- After the final rinse, add 70 kD of Texas Red-labeled dextran (0.6 mg/mL) and Hoechst (1:10,000) in 1x TRB-BSA. Leave the plate on the bench at room temperature for 15 min, protected from light.
- Image using a confocal microscope to identify the buffer and the BSA concentration in which the plasma membrane permeabilization percentage is the highest, but the nuclear membrane still restricts entry of the 70 kD dextran (goal ≥ 80%). Use these conditions for subsequent experiments.
4. Nuclear import assay
- Prepare the transport reactions
NOTE: To enable all reactions in the plate to be started simultaneously, prepare reaction mix in a plastic 96-well staging plate on ice, prior to neuron permeabilization. Carry out a minimum of two technical replicates per condition. Include controls on each plate.- Prepare the base reaction mix consisting of 2.5 mg/mL of WCE, 1x ERM, fluorescent cargo (200 nM Rango), and Hoechst 33342 (10 mg/mL, 1:10,000) in 1x TRB-BSA, sufficient for 50 µL per well. OPTION: Include 70 kD Texas red-labeled dextran to permit monitoring of nuclear pore integrity throughout the transport reaction, keeping in mind that confocal microscopy is needed to image nuclear dextran exclusion.
NOTE: The WCE concentration provided is a reasonable starting point for assay optimization. Perform empiric testing of each batch of WCE to determine the optimal concentration required for efficient nuclear import of the desired cargo. Avoid combining data from replicates prepared with different batches of WCE since import efficiency may vary. - Prepare the controls including (1) Cargo alone: Rango, but no WCE or ERM and (2) Inhibitor: fluorescent cargo, ERM, WCE, and 100 µM importazole (IPZ, a small molecule inhibitor of importin β18). As an alternate inhibitor, use 0.8 mg/mL of wheat germ agglutinin (WGA)19.
NOTE: Equilibrate the inhibitors in the assay mix containing WCE for at least 30 min before initiation of transport. For maximal inhibition, post-permeabilization rinse steps in corresponding wells should also include the inhibitor.
- Prepare the base reaction mix consisting of 2.5 mg/mL of WCE, 1x ERM, fluorescent cargo (200 nM Rango), and Hoechst 33342 (10 mg/mL, 1:10,000) in 1x TRB-BSA, sufficient for 50 µL per well. OPTION: Include 70 kD Texas red-labeled dextran to permit monitoring of nuclear pore integrity throughout the transport reaction, keeping in mind that confocal microscopy is needed to image nuclear dextran exclusion.
- Permeabilize and rinse the neurons
- Permeabilize the neurons according to the optimal, batch-specific protocol identified in step 3.
- Remove the culture plate from the incubator and immediately place it on ice. Rinse 2x for 5 min in 1x TRB-BSA. Include an inhibitor in the rinses for designated wells.
- Run the transport reaction.
- Following the final rinse, remove 1x TRB-BSA. Working quickly, use a multichannel pipette (50 µL/well) to transfer the premixed transport reactions onto the permeabilized cells.
- Immediately load the plate in a high content microscope at room temperature to initiate time-lapse imaging (see step 4.4) or place the plate on the bench, protected from light, and allow the reaction to proceed for the desired time period (e.g., 30-120 min) prior to fixation and imaging.
- If desired, fix in 4% paraformaldehyde/PBS for 15 min, rinse 2x for 5 min in PBS, and transfer to 50% glycerol/PBS for later imaging. Keep the fixed plates protected from light at 4 °C (stable for up to 1 week or more).
- Live imaging
NOTE: Apply any preferred image acquisition and analysis method at this stage.- Load the plate in a high content microscope. Run an initial test plate to optimize and save imaging parameters, including magnification, focus, exposure times (Hoechst and FITC), and intervals prior to running experimental samples. Save the parameters to reload with each subsequent plate.
NOTE: For quantitative analysis, set the exposure time for the fluorescent import cargo (FITC) well below saturation at steady state or the latest timepoint to be used. Establish this using the test plate, targeting half-maximal saturation to permit variations in intensity across experimental conditions. Collect the Hoechst images for each frame and timepoint to permit nuclear identification during subsequent automated analysis. - Adjust the focus offset, if needed, using Hoechst as the focus wavelength and run the imaging protocol. Collect the images every 5 min for 30 min. Adjust the interval and reaction time as needed based on the imaging speed and kinetics of the import reaction of interest.
- Load the plate in a high content microscope. Run an initial test plate to optimize and save imaging parameters, including magnification, focus, exposure times (Hoechst and FITC), and intervals prior to running experimental samples. Save the parameters to reload with each subsequent plate.
5. Image analysis
- In the microscope Review Plate dialog, open Hoechst and FITC images from a representative frame. Under Run Analysis tab, select Translocation-Enhanced module, and configure the settings. Set Hoechst as the compartment image and FITC as the translocation probe image.
- Under compartments, adjust approximate nuclear width, intensity above local background, and minimum/maximum area. Activate Auto Separate Touching Compartments as the neurons tend to cluster. View results by selecting Test Run. Adjust the settings to maximize the accuracy of neuronal nuclei identification while limiting erroneous inclusion of non-neuronal nuclei or debris.
NOTE: Glial contamination, if present, can typically be excluded at this step based on nuclear size. Determination of optimal compartment identification settings is a trade-off between accuracy and cell number and must be set empirically depending on the assay, cell type, and culture density. Stringent parameters will omit numerous cells but include fewer errors, while liberal parameters will be more inclusive but contain more errors. If different cell populations (i.e. disease mutants) or treatments are included that may affect nuclear size, we recommend setting size/intensity parameters that are broad enough to accommodate all conditions to avoid the need for multiple parameter sets within an experiment. - To avoid artifacts from the nuclear edge, define regions of measurement several pixels inside and outside the compartment edge defined by Hoechst staining. Depending on magnification and binning, set 2-4 pixels for inner region distance, and 1-2 pixels for outer region distance. Adjust the outer region width (1-2 pixels).
- Select the desired background estimation method (default of Auto Constant is recommended). In Configure data log, select the desired output parameters, including mean inner intensity, mean outer intensity, and inner/outer mean intensity (the nuclear to cytoplasmic (N/C) ratio). Save the analysis parameters.
NOTE: The inclusion of internal positive and negative controls within each experiment is strongly encouraged to ensure that parameters maximize sensitivity to detect the biological event of interest. - Use the Plate Utilities dialog to run analysis on the desired plate(s) using the saved parameters and export the measurements.
- Review and summarize the data by the treatment condition. OPTION: Apply filters to remove non-physiologic data, such as probe intensity = 0 and extremes of N/C ratio (i.e., <0.1 and >100). Equally apply any filters across all conditions and experiments.
- As an additional correction for baseline fluorescence, normalize the data to the control wells in which lysate and ER were omitted. To facilitate comparisons across biological replicates (i.e., independent neuronal culture preparations), express the final nuclear import data as a percent of time 0.
Representative Results
Selective permeabilization of the plasma membrane (Figure 1A) is the most critical step in the protocol and must be verified prior to proceeding with the analysis of nuclear import. Due to variations between each culture preparation, an initial titration plate is routinely run to identify the optimal, batch-specific concentrations of hypotonic Tris-HCl buffer and BSA cushion, as described in step 3. Under- and over-permeabilized cells are readily identified by confocal microscopy (Figure 1B) due to lack of penetration of 70 kD dextran into the cytoplasmic compartment or presence of dextran within both the cytoplasmic and nuclear compartments, respectively. In optimal conditions, the majority of cells (≥80%) show 70 kD dextran surrounding but not crossing the nuclear membrane.
Following optimization of permeabilization, the next critical phase, described in step 4, is to establish that nuclear import of fluorescent cargo is energy- and transport receptor-dependent (Figure 2A). Controls lacking ERM and WCE (source of importins and Ran cycle proteins) should show trace, if any, nuclear accumulation of fluorescent cargo (Figure 2B,C). Import should be blocked by the relevant nuclear import inhibitors. These controls must be validated for each cargo to verify that facilitated nuclear import is occurring by the expected mechanism. Once the nuclear transport assay is validated, experimental perturbations can be applied depending on the question of interest.
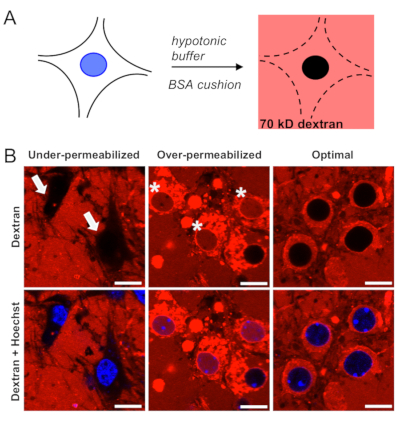
Figure 1: Selective permeabilization of the plasma membrane in mouse primary cortical neurons. (A) Schematic of neuronal permeabilization using the hypotonic Tris-HCl with BSA cushion, followed by validation with Texas red-labeled 70 kD dextran. (B) Confocal images of under-permeabilized neurons in which the plasma membrane remains intact, restricting entry of fluorescent dextran (arrows); over-permeabilized neurons in which dextran freely enters into the nucleus (asterisks); and optimally-permeabilized neurons in which dextran surrounds but does not enter the nucleus. Scale bars = 10 µm. Please click here to view a larger version of this figure.
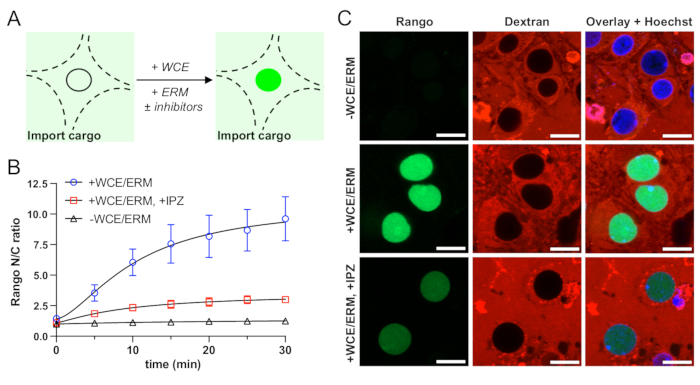
Figure 2: Validation of energy- and importin β-dependent nuclear import of Rango in permeabilized mouse primary cortical neurons. (A) Schematic of nuclear import reaction in permeabilized neurons. (B) Automated analysis of nuclear to cytoplasmic ratio (N/C) of Rango. Mean ± SEM is shown for n = 3 biological replicates (independent neuronal cultures), each containing two technical replicates (wells) per condition or approximately 400 neurons. (C) Confocal images of Rango nuclear import showing requirement for whole cell extract (WCE) and energy regeneration mix (ERM), and import blockade by importazole (IPZ, 100 µM). Confocal acquisition parameters were kept constant across all conditions. Scale bar = 10 µm. Please click here to view a larger version of this figure.
Discussion
The protocol detailed above provides a reliable and reproducible method for selectively permeabilizing the plasma membrane of primary mouse cortical neurons for nuclear import analysis. Here, an application of the method for nuclear import analysis of a direct importin β cargo (Rango) is shown, but this same approach can be used to analyze the passive and active import of a wide range of fluorescent cargoes. Permeabilization enables precise manipulation of the transport reaction in ways that are not feasible in intact cells, as we recently described for mutant C9orf72 dipeptide repeat proteins (DPRs) implicated in ALS and FTD14. Synthetic DPR proteins were preincubated with transport lysates prior to initiation of the nuclear import reaction, facilitating analysis of concentration- and length-dependence of the nuclear import blockade by arginine-containing DPRs. In fractionation studies, DPR-induced aggregates were included or excluded from the transport lysate to analyze functional consequences for nuclear import. DPRs were also preincubated with nuclei to ascertain effects on nuclear pore permeability. These are a few examples of variations of the assay that can be applied to the study of disordered and aggregation-prone proteins, commonly implicated in neurodegeneration. Since this is an artificial in vitro system, the findings should be carefully interpreted within the limitations of the assay.
Success of the protocol critically depends on selective permeabilization of the plasma membrane, to the extent that careful optimization of conditions for each batch of primary neurons is recommended. Importantly, this protocol is specific to primary mouse cortical neurons. We have also carried out preliminary successful testing with primary mouse spinal motor neurons, but caution that the hypotonic permeabilization method is not reliable in human-induced pluripotent stem cell-derived spinal neuron cultures (not shown). If active nuclear import is not observed, despite optimal permeabilization, troubleshooting efforts should focus on ensuring high quality, concentrated WCE and fluorescent cargo preparations. Due to the potential for batch-to-batch variation, it is best to prepare and freeze large stocks of single-use aliquots and avoid comparing results across different batches. The use of automated high content imaging and analysis permits rapid imaging of nuclear import kinetics across hundreds of neurons per well, with minimal investigator input. If high throughput imaging is not available, nuclear import reactions containing Rango and related cargoes are fixable14 for alternate imaging and analysis methods. If manual imaging and analysis are pursued, investigators should be blinded to treatment condition to avoid bias.
We anticipate that this method will be broadly applicable to studies of passive and active nuclear transport in primary neurons. As described above, permeabilized cells are a useful model for the study of the effect of disordered and aggregation-prone proteins, commonly implicated in neurodegeneration and increasingly suspected to disrupt nucleocytoplasmic transport20,21,22. Comparison between different cell types may also improve the understanding of potential differences in transport dynamics related to cell-specific differences in nuclear pore complex composition11,12. Thus far, no alteration in the concentrations of components was needed in the transport reaction mix for permeabilized neurons versus HeLa cells14. However, no formal comparisons were made of the minimal transport requirements and efficiency in permeabilized HeLa versus neurons or glia. In addition to varying the permeabilized cell type, utilization of neurons or even CNS tissue as the source of cytoplasmic or whole cell extract, thus varying the source of soluble transport factors, may provide an additional approach for testing disease-relevant questions.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by NINDS K08NS104273 (to L.R.H.).
Materials
| 1 M HEPES | Gibco | 15630-080 | |
| 10x HBSS | Gibco | 14185-052 | |
| 32% paraformaldehyde | Electron Microscopy Sciences | 15714-S | |
| 96-well optical glass plates | CellVis | P96-1.5H-N | |
| ATP lithium salt | Millipore Sigma | 11140965001 | |
| B27 | Gibco | 17504-044 | |
| Bio-Rad Protein Assay Kit II | Bio-Rad | 5000002 | |
| BL21(DE3) E. coli | NEB | C2527H | |
| Bovine serum albumin fraction V, heat shock, fatty acid free | Sigma-Aldrich | 3117057001 | |
| Chromatography columns | Bio-Rad | 7311550 | |
| Creatine kinase | Millipore Sigma | 10127566001 | |
| Creatine phosphate | Millipore Sigma | 10621722001 | |
| Dextran, Texas Red, 70,000 MW | Thermo Fisher | D1864 | |
| DNase I | Sigma-Aldrich | DN25 | |
| E15-16 timed pregnant C57BL/6J female mice | Jackson Laboratory | 000664 | |
| Excel | Microsoft | N/A | |
| Fetal bovine serum | Hyclone | SH30070.03 | |
| Glutamax | Gibco | 35050-061 | |
| Glycerol | Thermo Fisher | 15514011 | |
| GTP lithium salt | Millipore Sigma | 11140957001 | |
| HALT protease inhibitor (100x) | Thermo Fisher | 78439 | |
| HEK293T cells | ATCC | CRL-3216 | |
| HIS-Select HF Nickel affinity gel | Sigma-Aldrich | HO537 | |
| Hoechst 33342 | Thermo Fisher | H1399 | |
| ImageExpress Micro Confocal High-content Imaging System | Molecular Devices | N/A | Used for time-lapse imaging |
| Imidazole | Millipore | I3386 | |
| Importazole | Sigma-Aldrich | SML0341 | |
| IPTG | Corning | 46-102-RF | |
| Laminin | Sigma-Aldrich | L2020 | |
| LB broth | Grainger | 31FZ62 | |
| LSM800 confocal microscope | Zeiss | N/A | Used for dextran imaging |
| MetaXpress High Content Image Analysis Software | Molecular Devices | N/A | |
| Neurobasal medium | Gibco | 21103 | |
| Papain | Worthington | LS003126 | |
| Penicillin-streptomycin | Gibco | 15140-122 | |
| Poly-D-Lysine | Sigma-Aldrich | P6407 | |
| Protease inhibitor cocktail | Millipore Sigma | 11873580001 | |
| Rango Plasmid (pRSET Rango2/a1 + linkers) | N/A | N/A | pK44, containing N-terminal 6-His tag |
| SOC (super optimal broth with catabolite repression) media | Quality Biological | 340-031-671 | |
| Sodium pyruvate | Gibco | 11360-070 | |
| Spin-X UF concentrators (30K MWCO) | Corning | CLS431484 | |
| Trypsin-EDTA (0.05%) | Gibco | 25300054 |
References
- Hutten, S., Dormann, D. Nucleocytoplasmic transport defects in neurodegeneration – Cause or consequence. Seminars in Cell & Developmental Biology. 5, 151-162 (2019).
- Zhang, K., et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 525 (7567), 56-61 (2015).
- Grima, J. C., et al. Mutant Huntingtin disrupts the nuclear pore complex. Neuron. 94 (1), 93-96 (2017).
- Eftekharzadeh, B., et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron. 99 (5), 925-927 (2018).
- Coyne, A. N., et al. G4C2 repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron. 107 (6), 1124-1140 (2020).
- Timney, B. L., et al. Simple rules for passive diffusion through the nuclear pore complex. The Journal of Cell Biology. 215 (1), 57-76 (2016).
- Paci, G., Caria, J., Lemke, E. A. Cargo transport through the nuclear pore complex at a glance. Journal of Cell Science. 134 (2), 247874 (2021).
- Ding, B., Akter, M., Zhang, C. -. L. Differential Influence of Sample Sex and Neuronal Maturation on mRNA and Protein Transport in Induced Human Neurons. Frontiers in Molecular Neuroscience. 13, 46 (2020).
- Adam, S. A., Marr, R. S., Gerace, L. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. The Journal of Cell Biology. 111 (3), 807-816 (1990).
- Brownawell, A. M., Holaska, J. M., Macara, I. G., Paschal, B. M. The use of permeabilized cell systems to study nuclear transport. Methods in Molecular Biology. 189, 209-229 (2002).
- Raices, M., D’Angelo, M. A. Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nature Reviews. Molecular Cell Biology. 13 (11), 687-699 (2012).
- Ori, A., et al. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Molecular Systems Biology. 9, 648 (2013).
- Colbeau, A., Nachbaur, J., Vignais, P. M. Enzymic characterization and lipid composition of rat liver subcellular membranes. Biochimica et Biophysica Acta. 249 (2), 462-492 (1971).
- Hayes, L. R., Duan, L., Bowen, K., Kalab, P., Rothstein, J. D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife. 9, 51685 (2020).
- Grote, P., Ferrando-May, E. In vitro assay for the quantitation of apoptosis-induced alterations of nuclear envelope permeability. Nature Protocols. 1 (6), 3034-3040 (2006).
- Lowe, A. R., et al. Importin-β modulates the permeability of the nuclear pore complex in a Ran-dependent manner. eLife. 4, 04052 (2015).
- Kalab, P., Pralle, A., Isacoff, E. Y., Heald, R., Weis, K. Analysis of a RanGTP-regulated gradient in mitotic somatic cells. Nature. 440 (7084), 697-701 (2006).
- Soderholm, J. F., et al. Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chemical Biology. 6 (7), 700-708 (2011).
- Newmeyer, D. D., Forbes, D. J. Nuclear import can be separated into distinct steps in vitro: nuclear pore binding and translocation. Cell. 52 (5), 641-653 (1988).
- Woerner, A. C., et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 351 (6269), 173-176 (2016).
- Khosravi, B., et al. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Human Molecular Genetics. 26 (4), 790-800 (2016).
- Chou, C. -. C., et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nature Neuroscience. 21 (2), 228-239 (2018).

.