DFA results showed 100%, 100%, 83.3%, 66%, and 50% positivity for RABV in the cortex, thalamus, medulla, pons, and horn, respectively. These results confirmed the previous results, and at least three of the structures dissected from each brain were positive for RABV. A representative positive DAF staining is shown in Figure 1.
Figure 2 shows the amplification of a fragment of the RABV N gene (step 5.5) with the primers first ones reported by Loza-Rubio et al.11. This showed that the material obtained to be used in amplification was of good quality.
The size of the PCR-amplified fragment is shown in Figure 3. The equation of the standard curve shows the relationship between the amount of RABV genetic content in a sample and the size of the PCR-amplified fragment. The amount of RABV genetic content (in ng) was deduced by interpolation of the Ct values in the calibration curve using the equation presented in Figure 4. Using this equation, the detection limit of 1 x 105 μg/μL of viral RNA was found to correspond to 36 copies of the RABV genome. These results demonstrate that the assay is sensitive and can be used to detect the virus in samples that contain a low number of copies RABV N gene. The efficiency of the assay was calculated using the slope of the standard curve, which was -3.28. The samples used for qRT-PCR showed amplification of the RABV N gene.
To determine the number of viral copy present, the Ct values for each sample were interpolated using the calibration curve equation. The result obtained (in nanograms) was substituted into the formula described below from the following ref.24:
Number of copies RABV N gene = (ng sample * 6.022 x 1023) / (104 bp (length of PCR product) * 1 x 109 * 650).
Comparatively, the results of qRT-PCR assays were consistent with those obtained using DFA. According to the results obtained in the qRT-PCR test in cases C and D (Table 1), the thalamus is the structure that possesses the highest number of copies of the RABV N gene. This suggests that early infection of hypothalamic and thalamic neurons is important in the development of the disease, given that these neurons control the vegetative functions of an animal. The copy numbers of the RABV N gene that were detected in each structure of each sample are shown in Table 1. The sensitivity and specificity of the qRT-PCR test were both 100%, as all samples were positive. This result is consistent with the initial diagnoses of the samples.
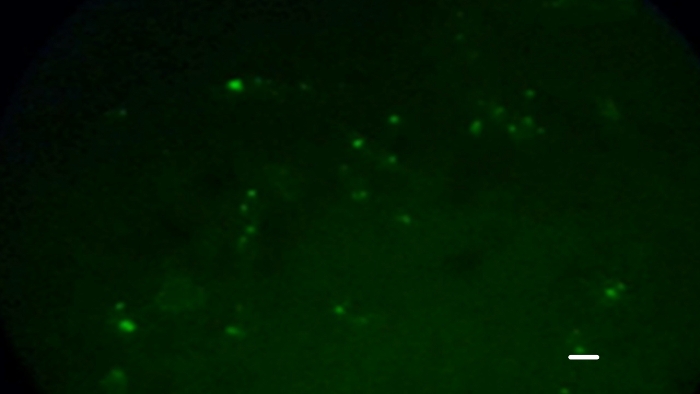
Figure 1: Bovine brain tissues showing positive staining according to the direct immunofluorescence test that detects rabies virus protein N in infected tissues. An apple green coloration is observed upon staining, where the antibody binds to the rabies antigen. Scale bar: 50 µm Please click here to view a larger version of this figure.
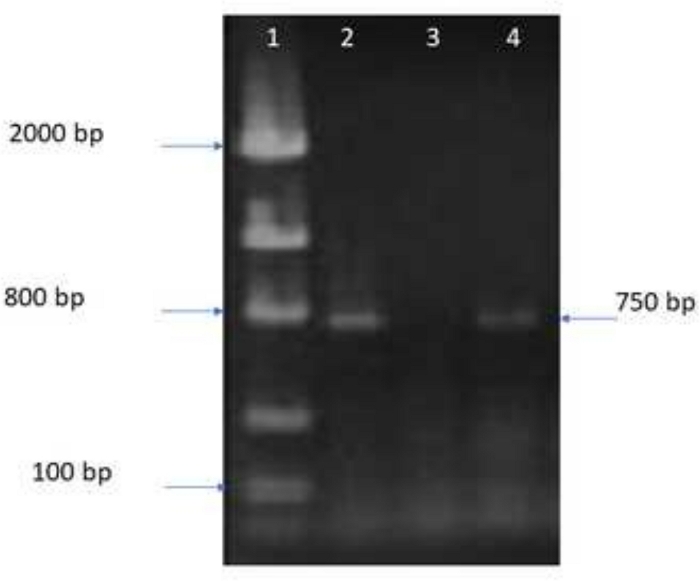
Figure 2: 1.5% agarose gel showing the amplification products. Amplification of a 761 bp fragment of the RABV N gene to demonstrate the presence and quality of the cDNA to be used in in vitro transcription. Lane 1 contains a molecular weight marker, line 2: amplification of positive control; lane 3: negative control (non-infected sample); line 4: amplification of cDNA used for in vitro transcription. Please click here to view a larger version of this figure.
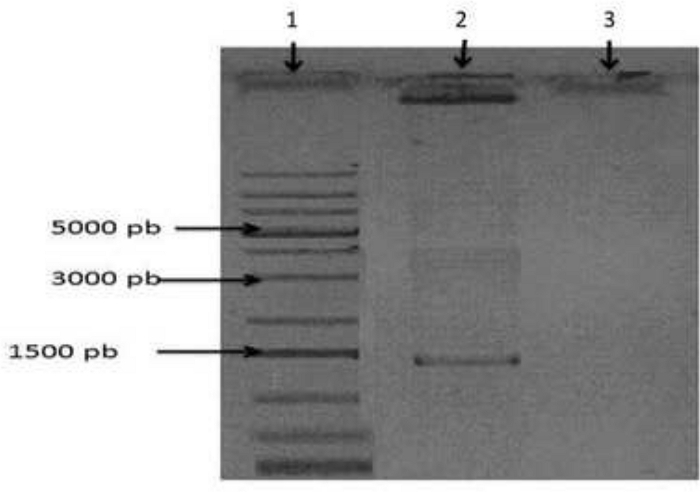
Figure 3: 1.5% agarose gel showing the amplification products. Amplification of the complete N gene is shown in lane 2. Lane 1 contains a molecular weight marker, and lane 3 contains the negative amplification control. Please click here to view a larger version of this figure.
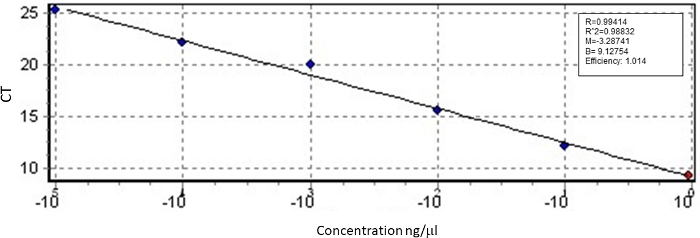
Figure 4: Standard curve of the qRT-PCR using in vitro transcribed N gene mRNA to quantify the number of copies of the RABV N gene. Please click here to view a larger version of this figure.
| ID brain | Structure | DAF | Number copies (x109) |
| A | Ammon’s horn | + | 0.261 |
| Cerebellum | + | 0.0663 | |
| Cortex | + | 0.02 | |
| Medulla | + | 0.146 | |
| Pons | + | 30.7 | |
| Thalamus | + | 108 | |
| B | Ammon’s horn | – | 0.0251 |
| Cerebellum | + | 4.64 | |
| Cortex | + | 12.4 | |
| Medulla | + | 0.175 | |
| Pons | ‒ | 0.0721 | |
| Thalamus | + | 121 | |
| C | Ammon’s horn | ‒ | 0.168 |
| Cerebellum | + | 0.0239 | |
| Cortex | + | 21.5 | |
| Medulla | + | 17.2 | |
| Pons | + | 1.32 | |
| Thalamus | + | 102 | |
| D | Ammon’s horn | + | 11.8 |
| Cerebellum | + | 0.0239 | |
| Cortex | + | 8.72 | |
| Medulla | + | 1.25 | |
| Pons | ‒ | 0.0165 | |
| Thalamus | + | 33.4 | |
| E | Ammon’s horn | + | 4.15 |
| Cerebellum | + | 6.78 | |
| Cortex | + | 1.53 | |
| Medulla | + | 1.41 | |
| Pons | + | 0.025 | |
| Thalamus | + | 95 | |
| F | Ammon’s horn | + | 0.0221 |
| Cerebellum | ‒ | 0.00023 | |
| Cortex | + | 4.95 | |
| Medulla | ‒ | 0.000556 | |
| Pons | + | 1.02 | |
| Thalamus | + | 89 |
Table 1: Determination of the number of copies of the RABV N gene within each brain structure. Results were obtained from six anatomical structures of RABV-positive bovine brains diagnosed using DFA. The words highlighted in bold indicate the structures with the most copies. The results were expressed as number of copies of the N gene per milligram of tissue.
